

Multiple Motif Scanning to identify methyltransferases from the yeast proteome
- PMID: 19351663
- PMCID: PMC2709183
- DOI: 10.1074/mcp.M900025-MCP200
Multiple Motif Scanning to identify methyltransferases from the yeast proteome
Abstract
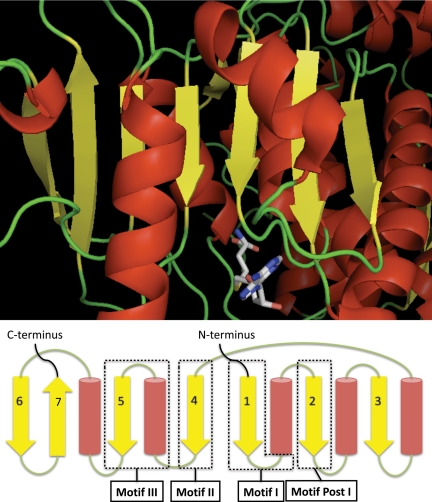
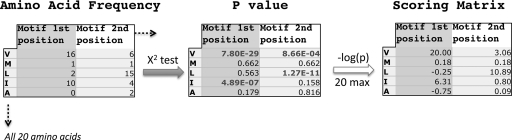
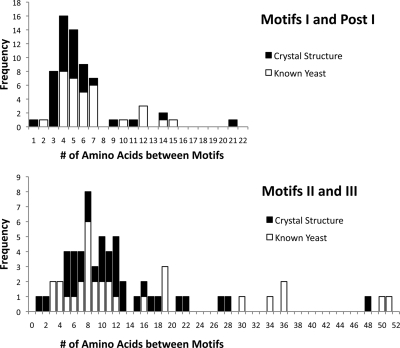
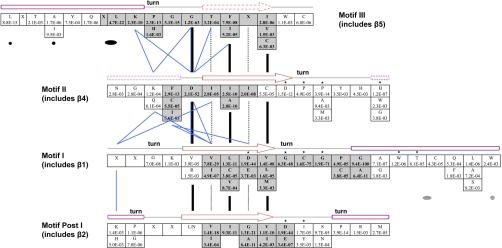
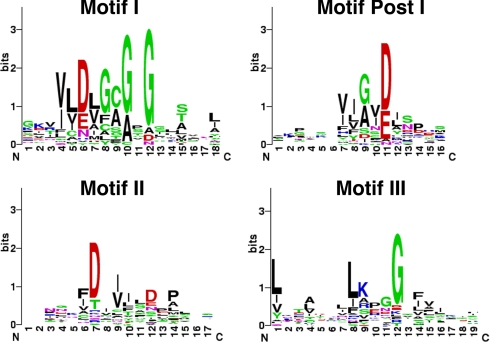
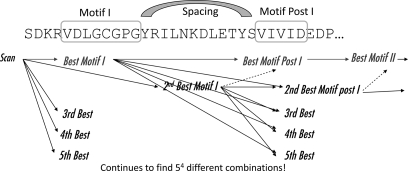
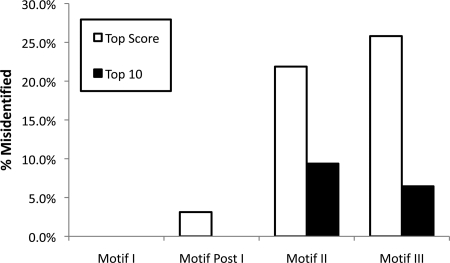
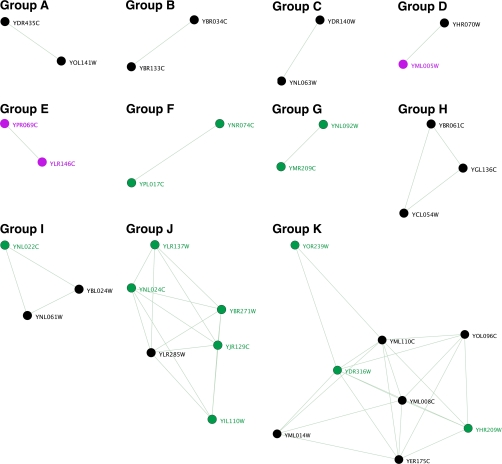
A new program (Multiple Motif Scanning) was developed to scan the Saccharomyces cerevisiae proteome for Class I S-adenosylmethionine-dependent methyltransferases. Conserved Motifs I, Post I, II, and III were identified and expanded in known methyltransferases by primary sequence and secondary structural analysis through hidden Markov model profiling of both a yeast reference database and a reference database of methyltransferases with solved three-dimensional structures. The roles of the conserved amino acids in the four motifs of the methyltransferase structure and function were then analyzed to expand the previously defined motifs. Fisher-based negative log statistical matrix sets were developed from the prevalence of amino acids in the motifs. Multiple Motif Scanning is able to scan the proteome and score different combinations of the top fitting sequences for each motif. In addition, the program takes into account the conserved number of amino acids between the motifs. The output of the program is a ranked list of proteins that can be used to identify new methyltransferases and to reevaluate the assignment of previously identified putative methyltransferases. The Multiple Motif Scanning program can be used to develop a putative list of enzymes for any type of protein that has one or more motifs conserved at variable spacings and is freely available (www.chem.ucla.edu/files/MotifSetup.Zip). Finally hidden Markov model profile clustering analysis was used to subgroup Class I methyltransferases into groups that reflect their methyl-accepting substrate specificity.
Figures

Similar articles
-
Widespread occurrence of three sequence motifs in diverse S-adenosylmethionine-dependent methyltransferases suggests a common structure for these enzymes.Arch Biochem Biophys. 1994 May 1;310(2):417-27. doi: 10.1006/abbi.1994.1187. Arch Biochem Biophys. 1994. PMID: 8179327
-
Uncovering the human methyltransferasome.Mol Cell Proteomics. 2011 Jan;10(1):M110.000976. doi: 10.1074/mcp.M110.000976. Epub 2010 Oct 7. Mol Cell Proteomics. 2011. PMID: 20930037 Free PMC article.
-
Automated identification of putative methyltransferases from genomic open reading frames.Mol Cell Proteomics. 2003 Aug;2(8):525-40. doi: 10.1074/mcp.M300037-MCP200. Epub 2003 Jul 18. Mol Cell Proteomics. 2003. PMID: 12872006
-
Five hierarchical levels of sequence-structure correlation in proteins.Appl Bioinformatics. 2004;3(2-3):97-104. doi: 10.2165/00822942-200403020-00004. Appl Bioinformatics. 2004. PMID: 15693735 Review.
-
The phospholipid methyltransferases in yeast.Biochim Biophys Acta. 1997 Sep 4;1348(1-2):134-41. doi: 10.1016/s0005-2760(97)00121-5. Biochim Biophys Acta. 1997. PMID: 9370325 Review.
Cited by
-
A novel small molecule methyltransferase is important for virulence in Candida albicans.ACS Chem Biol. 2013 Dec 20;8(12):2785-93. doi: 10.1021/cb400607h. Epub 2013 Oct 16. ACS Chem Biol. 2013. PMID: 24083538 Free PMC article.
-
A novel automethylation reaction in the Aspergillus nidulans LaeA protein generates S-methylmethionine.J Biol Chem. 2013 May 17;288(20):14032-14045. doi: 10.1074/jbc.M113.465765. Epub 2013 Mar 26. J Biol Chem. 2013. PMID: 23532849 Free PMC article.
-
Closing in on human methylation-the versatile family of seven-β-strand (METTL) methyltransferases.Nucleic Acids Res. 2024 Oct 28;52(19):11423-11441. doi: 10.1093/nar/gkae816. Nucleic Acids Res. 2024. PMID: 39351878 Free PMC article. Review.
-
Probabilistic approach to predicting substrate specificity of methyltransferases.PLoS Comput Biol. 2014 Mar 20;10(3):e1003514. doi: 10.1371/journal.pcbi.1003514. eCollection 2014 Mar. PLoS Comput Biol. 2014. PMID: 24651469 Free PMC article.
-
Emerging technologies to map the protein methylome.J Mol Biol. 2014 Oct 9;426(20):3350-62. doi: 10.1016/j.jmb.2014.04.024. Epub 2014 May 5. J Mol Biol. 2014. PMID: 24805349 Free PMC article. Review.
References
-
- Cheng X., Blumenthal R. M. ( 1999) S-Adenosylmethionine-Dependent Methyltransferases: Structures and Functions, World Scientific, Singapore
-
- Djordjevic S., Stock A. M. ( 1997) Crystal structure of the chemotaxis receptor methyltransferase CheR suggests a conserved structural motif for binding S-adenosylmethionine. Structure 5, 545– 558 - PubMed
-
- Martin J. L., McMillan F. M. ( 2002) SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12, 783– 793 - PubMed
-
- Schluckebier G., O'Gara M., Saenger W, Cheng X. ( 1995) Universal catalytic domain structure of AdoMet-dependent methyltransferases. J. Mol. Biol. 247, 16– 20 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
