Cross species analysis of microarray expression data
- PMID: 19357096
- PMCID: PMC2732912
- DOI: 10.1093/bioinformatics/btp247
Cross species analysis of microarray expression data
Abstract
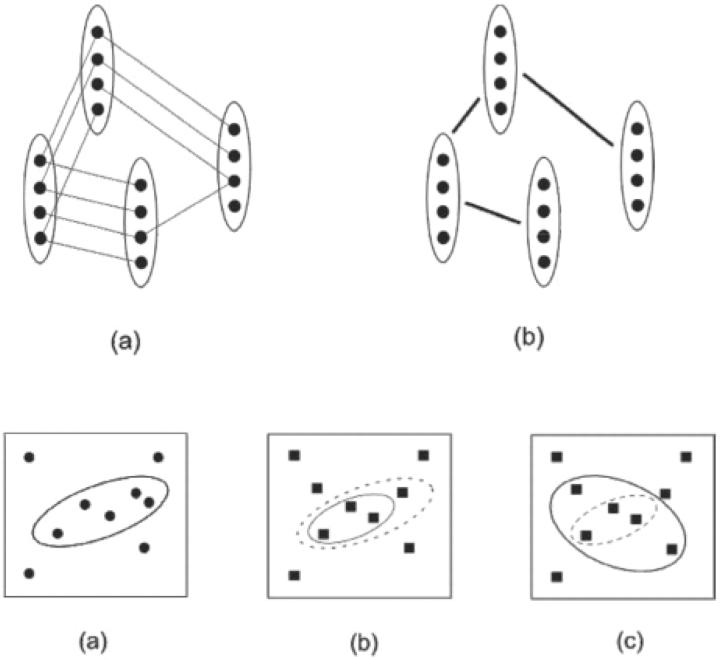
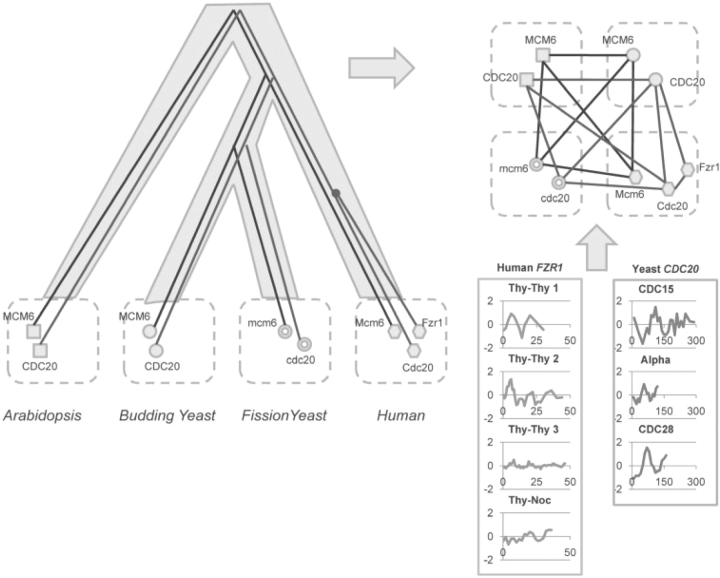
Motivation: Many biological systems operate in a similar manner across a large number of species or conditions. Cross-species analysis of sequence and interaction data is often applied to determine the function of new genes. In contrast to these static measurements, microarrays measure the dynamic, condition-specific response of complex biological systems. The recent exponential growth in microarray expression datasets allows researchers to combine expression experiments from multiple species to identify genes that are not only conserved in sequence but also operated in a similar way in the different species studied.
Results: In this review we discuss the computational and technical challenges associated with these studies, the approaches that have been developed to address these challenges and the advantages of cross-species analysis of microarray data. We show how successful application of these methods lead to insights that cannot be obtained when analyzing data from a single species. We also highlight current open problems and discuss possible ways to address them.
Figures