

Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease
- PMID: 19358844
- PMCID: PMC2710427
- DOI: 10.1016/j.expneurol.2009.03.042
Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer's disease
Abstract
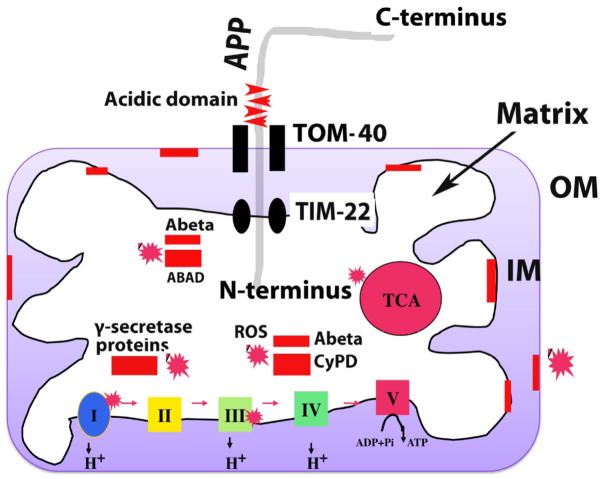
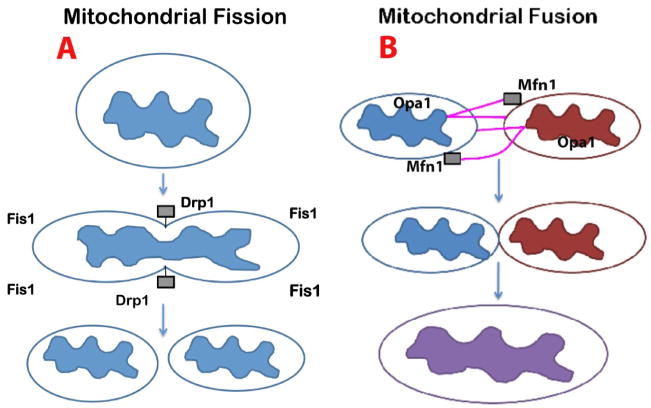
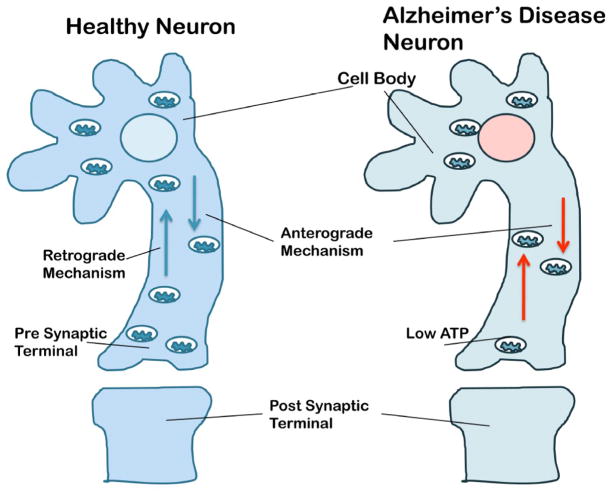
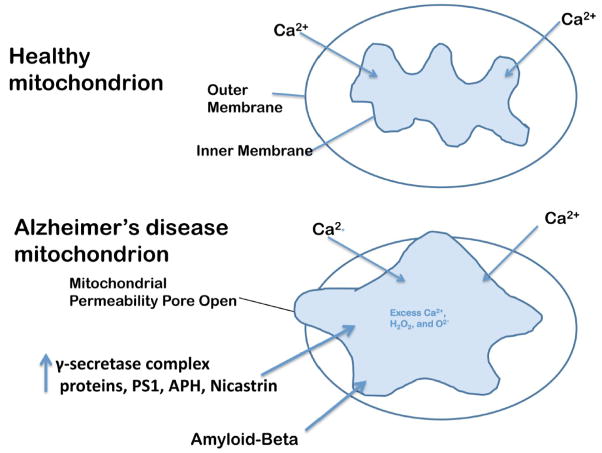
Mitochondria are the major source of energy for the normal functioning of brain cells. Increasing evidence suggests that the amyloid precursor protein (APP) and amyloid beta (Abeta) accumulate in mitochondrial membranes, cause mitochondrial structural and functional damage, and prevent neurons from functioning normally. Oligomeric Abeta is reported to induce intracellular Ca(2+) levels and to promote the excess accumulation of intracellular Ca(2+) into mitochondria, to induce the mitochondrial permeability transition pore to open, and to damage mitochondrial structure. Based on recent gene expression studies of APP transgenic mice and AD postmortem brains, and APP/Abeta and mitochondrial structural studies, we propose that the overexpression of APP and the increased production of Abeta may cause structural changes of mitochondria, including an increase in the production of defective mitochondria, a decrease in mitochondrial trafficking, and the alteration of mitochondrial dynamics in neurons affected by AD. This article discusses some critical issues of APP/Abeta associated with mitochondria, mitochondrial structural and functional damage, and abnormal intracellular calcium regulation in neurons from AD patients. This article also discusses the link between Abeta and impaired mitochondrial dynamics in AD.
Figures
References
-
- Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. - PubMed
-
- Anandatheerthavarada HK, Devi L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist. 2007 Dec;13:626–638. - PubMed
-
- Baloyannis SJ, Mauroudis I, Manolides SL, Manolides LS. Synaptic alterations in the medial geniculate bodies and the inferior colliculi in Alzheimer's disease: a Golgi and electron microscope study. Acta Otolaryngol. 2008;30:1–3. - PubMed
-
- Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T. Rossignol R.Mitochondrial bioenergetics and structural network organization. J Cell Sci. 2007;120:838–848. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
