

Topologies of complexes containing O6-alkylguanine-DNA alkyltransferase and DNA
- PMID: 19358853
- PMCID: PMC2736636
- DOI: 10.1016/j.jmb.2009.03.067
Topologies of complexes containing O6-alkylguanine-DNA alkyltransferase and DNA
Abstract
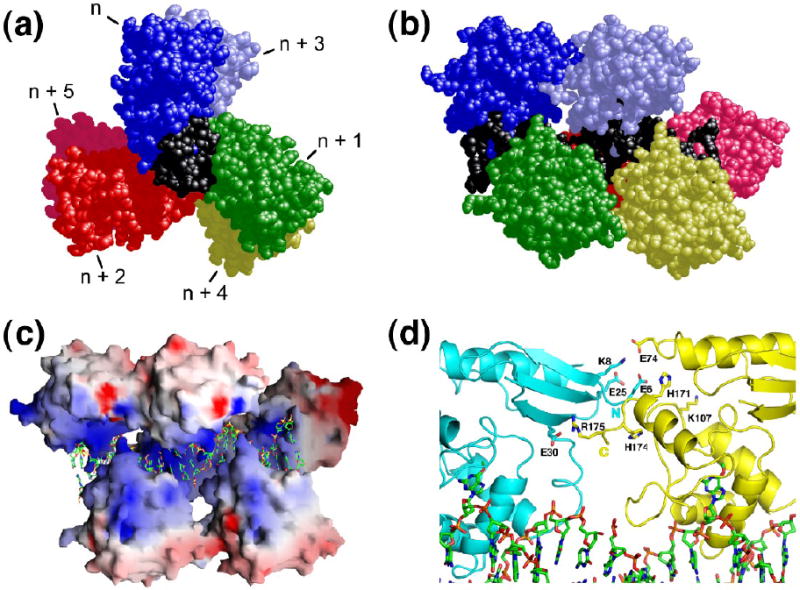
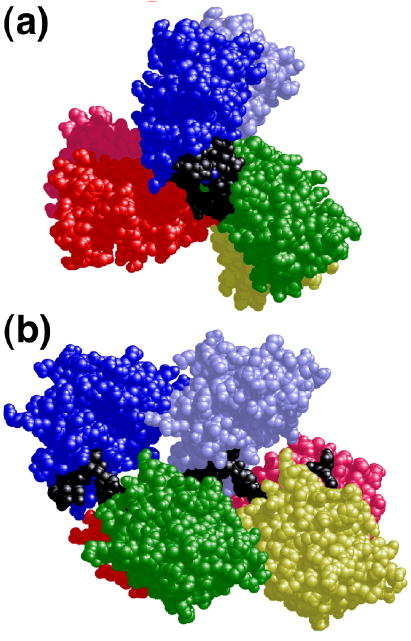
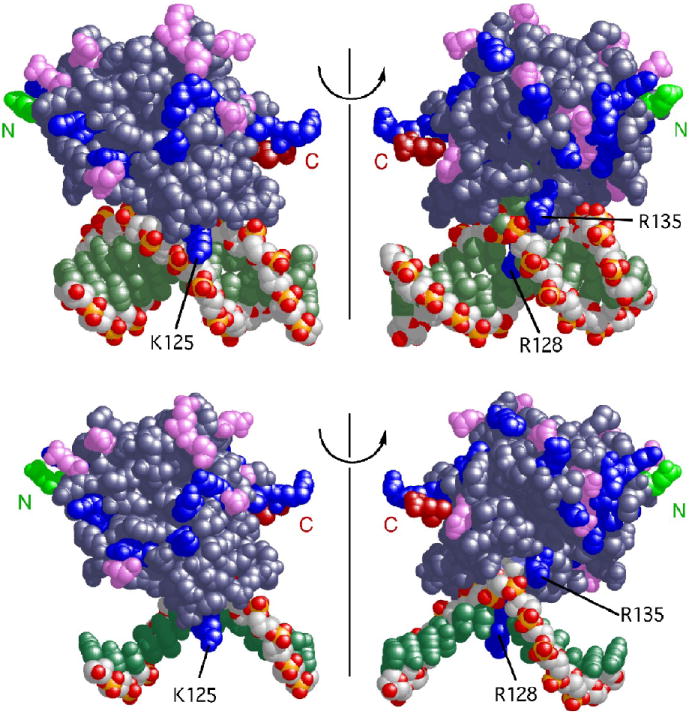
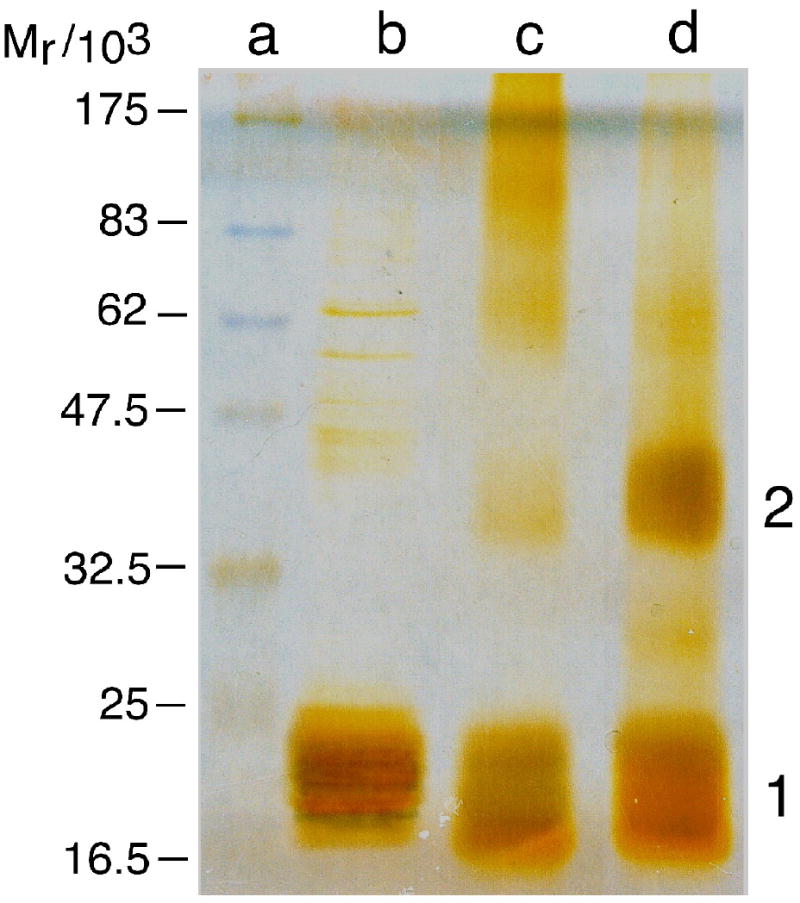
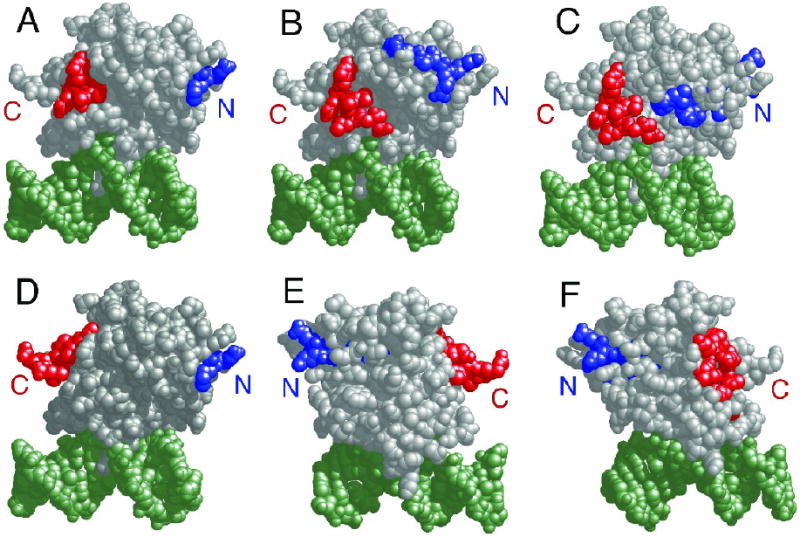
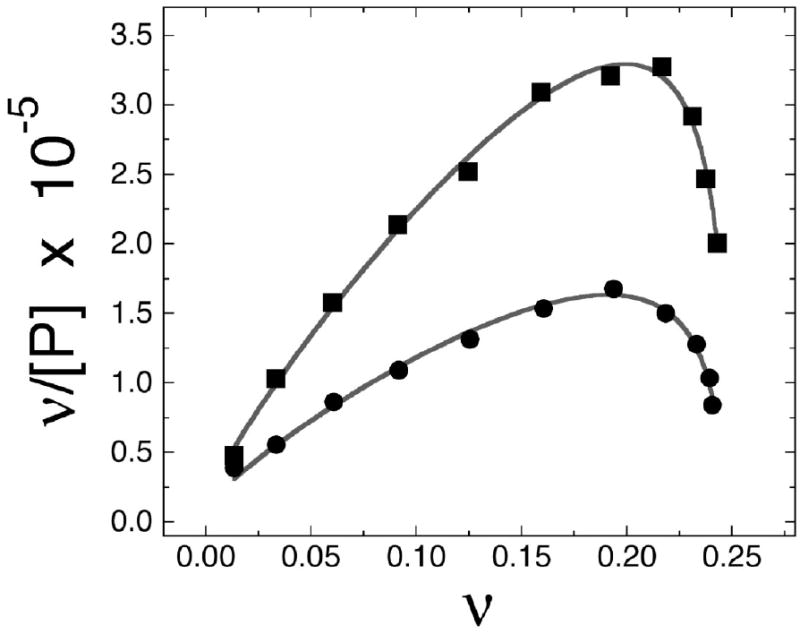
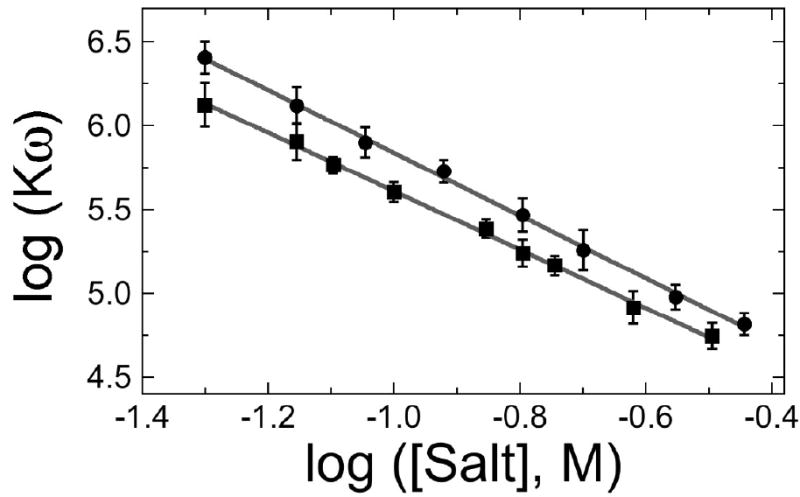
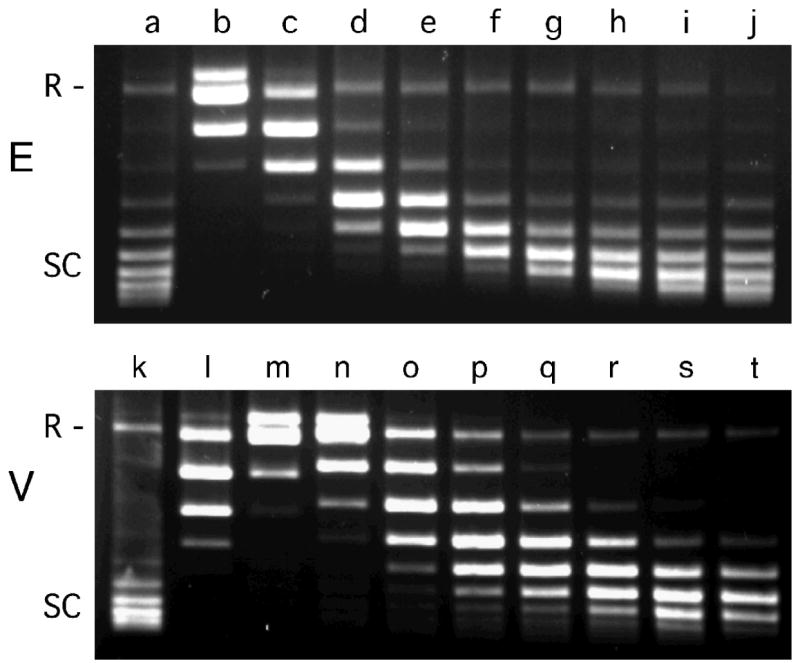
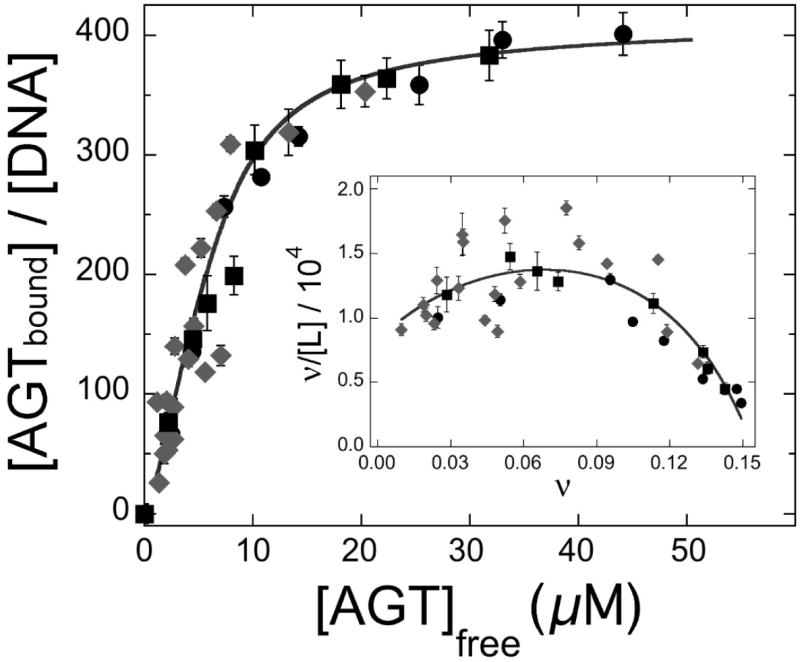
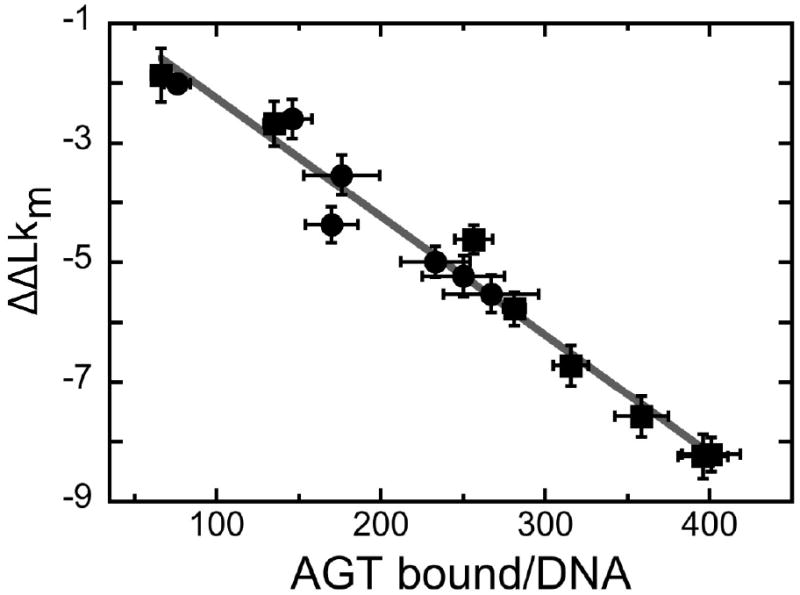
The mutagenic and cytotoxic effects of many alkylating agents are reduced by O(6)-alkylguanine-DNA alkyltransferase (AGT). In humans, this protein not only protects the integrity of the genome, but also contributes to the resistance of tumors to DNA-alkylating chemotherapeutic agents. Here we describe and test models for cooperative multiprotein complexes of AGT with single-stranded and duplex DNAs that are based on in vitro binding data and the crystal structure of a 1:1 AGT-DNA complex. These models predict that cooperative assemblies contain a three-start helical array of proteins with dominant protein-protein interactions between the amino-terminal face of protein n and the carboxy-terminal face of protein n+3, and they predict that binding duplex DNA does not require large changes in B-form DNA geometry. Experimental tests using protein cross-linking analyzed by mass spectrometry, electrophoretic and analytical ultracentrifugation binding assays, and topological analyses with closed circular DNA show that the properties of multiprotein AGT-DNA complexes are consistent with these predictions.
Figures






References
-
- Loveless A. Possible relevance of O-6 alkylation of dexoyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature (London) 1969;223:206–207. - PubMed
-
- Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogens and therapeutic agents. Cancer Res. 1990;50:6119–6129. - PubMed
-
- Margison GP, Santibáñez-Koref MF. O6-Alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. BioEssays. 2002;24:255–266. - PubMed
-
- Snow ET, Mitra S. Do carcinogen-modified deoxynucleotide precursors contribute to cellular mutagenesis? Cancer Invest. 1987;5:119–125. - PubMed
-
- Encell LP, Loeb lA. Enhanced in vivo repair of O4-methylthymine by a mutant human DNA alkyltransferase. Carcinogenesis. 2000;21:1397–1402. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RR-20171/RR/NCRR NIH HHS/United States
- R01 NS038041/NS/NINDS NIH HHS/United States
- NS-38041/NS/NINDS NIH HHS/United States
- DA-02243/DA/NIDA NIH HHS/United States
- P20 RR020171/RR/NCRR NIH HHS/United States
- GM-070662/GM/NIGMS NIH HHS/United States
- R01 DA002243/DA/NIDA NIH HHS/United States
- T32 GM008601/GM/NIGMS NIH HHS/United States
- R56 NS038041/NS/NINDS NIH HHS/United States
- R37 CA018137/CA/NCI NIH HHS/United States
- CA-018137/CA/NCI NIH HHS/United States
- R01 CA018137/CA/NCI NIH HHS/United States
- R01 CA097209/CA/NCI NIH HHS/United States
- CA-097209/CA/NCI NIH HHS/United States
- R01 GM070662/GM/NIGMS NIH HHS/United States
- 5 T32 GM-08601-05/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
