

CCN3 (NOV) is a negative regulator of CCN2 (CTGF) and a novel endogenous inhibitor of the fibrotic pathway in an in vitro model of renal disease
- PMID: 19359517
- PMCID: PMC2671261
- DOI: 10.2353/ajpath.2009.080241
CCN3 (NOV) is a negative regulator of CCN2 (CTGF) and a novel endogenous inhibitor of the fibrotic pathway in an in vitro model of renal disease
Abstract
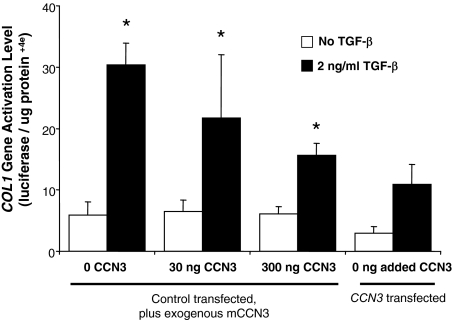
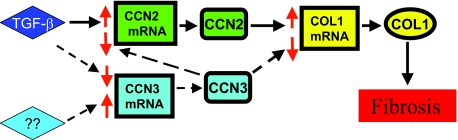
Fibrosis is a major cause of end-stage renal disease, and although initiation factors have been elucidated, uncertainty concerning the downstream pathways has hampered the development of anti-fibrotic therapies. CCN2 (CTGF) functions downstream of transforming growth factor (TGF)-beta, driving increased extracellular matrix (ECM) accumulation and fibrosis. We examined the possibility that CCN3 (NOV), another CCN family member with reported biological activities that differ from CCN2, might act as an endogenous negative regulator of ECM and fibrosis. We show that cultured rat mesangial cells express CCN3 mRNA and protein, and that TGF-beta treatment reduced CCN3 expression levels while increasing CCN2 and collagen type I activities. Conversely, either the addition of CCN3 or CCN3 overexpression produced a marked down-regulation of CCN2 followed by virtual blockade of both collagen type I transcription and its accumulation. This finding occurred in both growth-arrested and CCN3-transfected cells under normal growth conditions after TGF-beta treatment. These effects were not attributable to altered cellular proliferation as determined by cell cycle analysis, nor were they attributable to interference of Smad signaling as shown by analysis of phosphorylated Smad3 levels. In conclusion, both CCN2 and CCN3 appear to act in a yin/yang manner to regulate ECM metabolism. CCN3, acting downstream of TGF-beta to block CCN2 and the up-regulation of ECM, may therefore serve to naturally limit fibrosis in vivo and provide opportunities for novel, endogenous-based therapeutic treatments.
Figures








References
-
- Perbal B. CCN proteins: multifunctional signalling regulators. Lancet. 2004;363:62–64. - PubMed
-
- Rachfal AW, Brigstock DR. Structural and functional properties of CCN proteins. Vitam Horm. 2005;70:69–103. - PubMed
-
- Riser BL, Denichilo M, Cortes P, Baker C, Grondin JM, Yee J, Narins RG. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol. 2000;11:25–38. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
