

IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response
- PMID: 19361614
- PMCID: PMC2667992
- DOI: 10.1016/j.ajhg.2009.03.014
IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response
Abstract
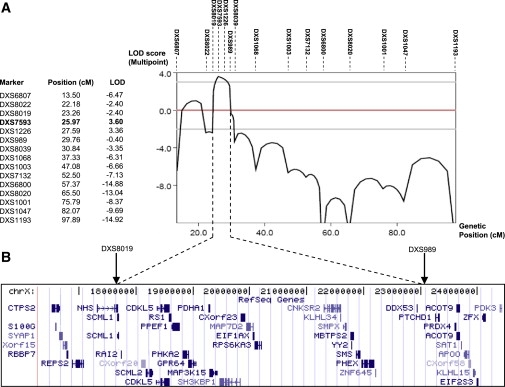
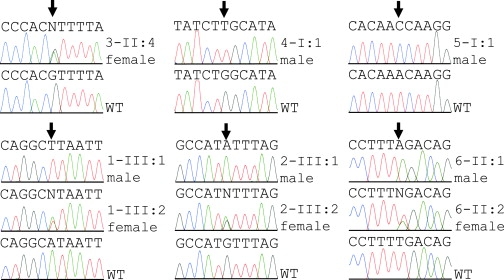
Ichthyosis follicularis with atrichia and photophobia (IFAP syndrome) is a rare X-linked, oculocutaneous human disorder. Here, we assign the IFAP locus to the 5.4 Mb region between DXS989 and DXS8019 on Xp22.11-p22.13 and provide evidence that missense mutations exchanging highly conserved amino acids of membrane-bound transcription factor protease, site 2 (MBTPS2) are associated with this phenotype. MBTPS2, a membrane-embedded zinc metalloprotease, activates signaling proteins involved in sterol control of transcription and ER stress response. Wild-type MBTPS2 was able to complement the protease deficiency in Chinese hamster M19 cells as shown by induction of an SRE-regulated reporter gene in transient transfection experiments and by growth of stably transfected cells in media devoid of cholesterol and lipids. These functions were impaired in five mutations as detected in unrelated patients. The degree of diminished activity correlated with clinical severity as noted in male patients. Our findings indicate that the phenotypic expression of IFAP syndrome is quantitatively related to a reduced function of a key cellular regulatory system affecting cholesterol homeostasis and ability to cope with ER stress.
Figures



References
-
- Macleod J.M.H. Three cases of “ichthyosis follicularis” associated with baldness. Br. J. Dermatol. 1909;21:165–189.
-
- Boente M.C., Bibas-Bonet H., Coronel A.M., Asial R.A. Atrichia, ichthyosis, follicular hyperkeratosis, chronic candidiasis, keratitis, seizures, mental retardation and inguinal hernia: A severe manifestation of IFAP syndrome? Eur. J. Dermatol. 2000;10:98–102. - PubMed
-
- Sato-Matsumura K.C., Matsumura T., Kumakiri M., Hosokawa K., Nakamura H., Kobayashi H., Ohkawara A. Ichthyosis follicularis with alopecia and photophobia in a mother and daughter. Br. J. Dermatol. 2000;142:157–162. - PubMed
-
- Cambiaghi S., Barbareschi M., Tadini G. Ichthyosis follicularis with atrichia and photophobia (IFAP) syndrome in two unrelated female patients. J. Am. Acad. Dermatol. 2002;46:S156–S158. - PubMed
-
- Khandpur S., Bhat R., Ramam M. Ichthyosis follicularis, alopecia and photophobia (IFAP) syndrome treated with acitretin. J. Eur. Acad. Dermatol. Venereol. 2005;19:759–762. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
