

Identification of potential driver genes in human liver carcinoma by genomewide screening
- PMID: 19366792
- PMCID: PMC2750086
- DOI: 10.1158/0008-5472.CAN-09-0164
Identification of potential driver genes in human liver carcinoma by genomewide screening
Abstract
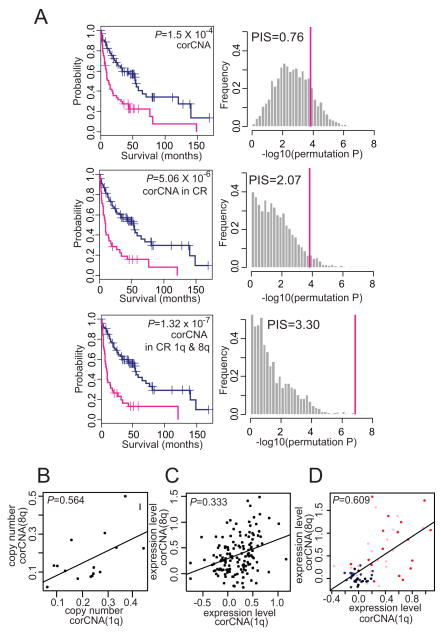
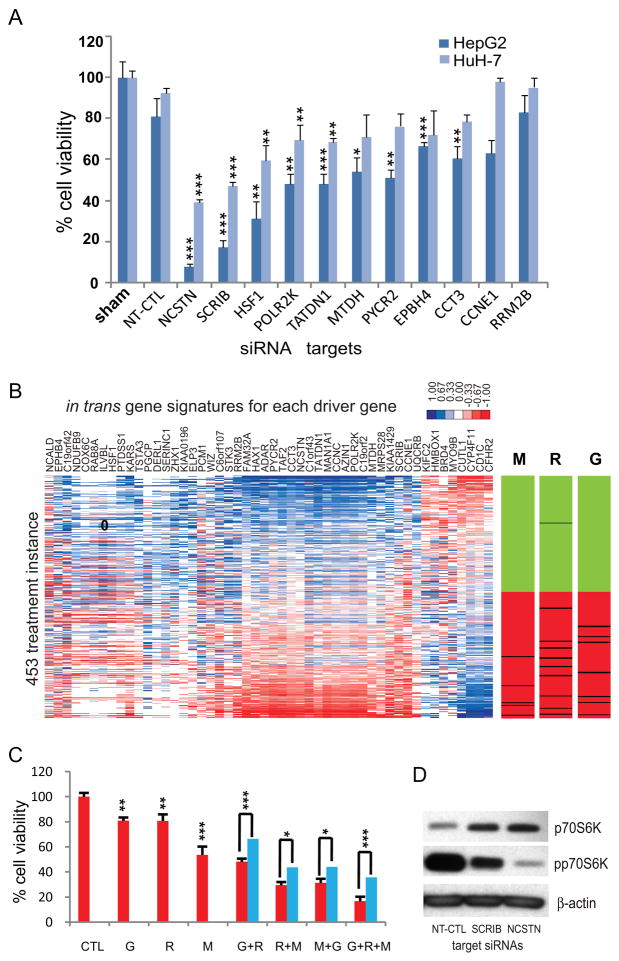
Genomic copy number aberrations and corresponding transcriptional deregulation in the cancer genome have been suggested to have regulatory roles in cancer development and progression. However, functional evaluation of individual genes from lengthy lists of candidate genes from genomic data sets presents a significant challenge. Here, we report effective gene selection strategies to identify potential driver genes based on systematic integration of genome scale data of DNA copy numbers and gene expression profiles. Using regional pattern recognition approaches, we discovered the most probable copy number-dependent regions and 50 potential driver genes. At each step of the gene selection process, the functional relevance of the selected genes was evaluated by estimating the prognostic significance of the selected genes. Further validation using small interference RNA-mediated knockdown experiments showed proof-of-principle evidence for the potential driver roles of the genes in hepatocellular carcinoma progression (i.e., NCSTN and SCRIB). In addition, systemic prediction of drug responses implicated the association of the 50 genes with specific signaling molecules (mTOR, AMPK, and EGFR). In conclusion, the application of an unbiased and integrative analysis of multidimensional genomic data sets can effectively screen for potential driver genes and provides novel mechanistic and clinical insights into the pathobiology of hepatocellular carcinoma.
Figures


References
-
- Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–76. - PubMed
-
- Wurmbach E, Chen YB, Khitrov G, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45:938–47. - PubMed
-
- Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9:416–23. - PubMed
-
- Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–6. - PubMed
-
- Paris PL, Andaya A, Fridlyand J, et al. Whole genome scanning identifies genotypes associated with recurrence and metastasis in prostate tumors. Hum Mol Genet. 2004;13:1303–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
