

Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy
- PMID: 19366804
- PMCID: PMC2765801
- DOI: 10.1158/0008-5472.CAN-08-3605
Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy
Abstract
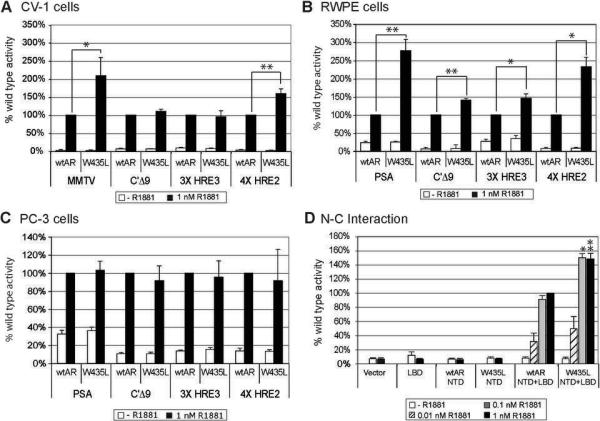
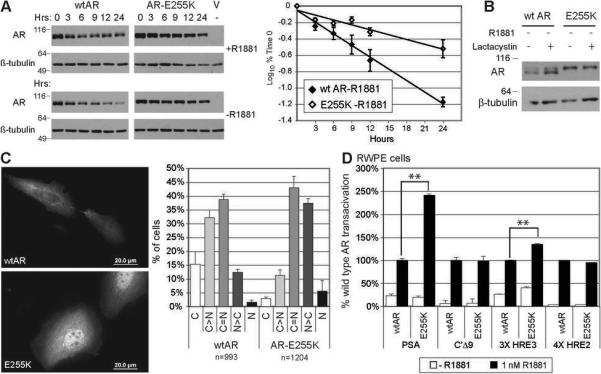
Mutations in the androgen receptor (AR) that enable activation by antiandrogens occur in hormone-refractory prostate cancer, suggesting that mutant ARs are selected by treatment. To validate this hypothesis, we compared AR variants in metastases obtained by rapid autopsy of patients treated with flutamide or bicalutamide, or by excision of lymph node metastases from hormone-naïve patients. AR mutations occurred at low levels in all specimens, reflecting genetic heterogeneity of prostate cancer. Base changes recurring in multiple samples or multiple times per sample were considered putative selected mutations. Of 26 recurring missense mutations, most in the NH(2)-terminal domain (NTD) occurred in multiple tumors, whereas those in the ligand binding domain (LBD) were case specific. Hormone-naïve tumors had few recurring mutations and none in the LBD. Several AR variants were assessed for mechanisms that might underlie treatment resistance. Selection was evident for the promiscuous receptor AR-V716M, which dominated three metastases from one flutamide-treated patient. For the inactive cytoplasmically restricted splice variant AR23, coexpression with AR enhanced ligand response, supporting a decoy function. A novel NTD mutation, W435L, in a motif involved in intramolecular interaction influenced promoter-selective, cell-dependent transactivation. AR-E255K, mutated in a domain that interacts with an E3 ubiquitin ligase, led to increased protein stability and nuclear localization in the absence of ligand. Thus, treatment with antiandrogens selects for gain-of-function AR mutations with altered stability, promoter preference, or ligand specificity. These processes reveal multiple targets for effective therapies regardless of AR mutation.
Figures


Similar articles
-
Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist.Cancer Res. 1999 Jun 1;59(11):2511-5. Cancer Res. 1999. PMID: 10363963
-
Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome.Cancer Res. 2003 Jan 1;63(1):149-53. Cancer Res. 2003. PMID: 12517791
-
Profiling human androgen receptor mutations reveals treatment effects in a mouse model of prostate cancer.Mol Cancer Res. 2008 Nov;6(11):1691-701. doi: 10.1158/1541-7786.MCR-08-0273. Mol Cancer Res. 2008. PMID: 19010817 Free PMC article.
-
Androgen receptor as a target in androgen-independent prostate cancer.Urology. 2002 Sep;60(3 Suppl 1):132-8; discussion 138-9. doi: 10.1016/s0090-4295(02)01593-5. Urology. 2002. PMID: 12231070 Review.
-
Targeting the androgen receptor in prostate cancer.Expert Opin Pharmacother. 2014 Jul;15(10):1427-37. doi: 10.1517/14656566.2014.915313. Epub 2014 Jun 2. Expert Opin Pharmacother. 2014. PMID: 24890318 Review.
Cited by
-
Androgen receptor gene rearrangements: new perspectives on prostate cancer progression.Curr Drug Targets. 2013 Apr;14(4):441-9. doi: 10.2174/1389450111314040005. Curr Drug Targets. 2013. PMID: 23410127 Free PMC article. Review.
-
The role of intracrine androgen metabolism, androgen receptor and apoptosis in the survival and recurrence of prostate cancer during androgen deprivation therapy.Curr Drug Targets. 2013 Apr;14(4):420-40. doi: 10.2174/1389450111314040004. Curr Drug Targets. 2013. PMID: 23565755 Free PMC article. Review.
-
New Opportunities for Targeting the Androgen Receptor in Prostate Cancer.Cold Spring Harb Perspect Med. 2018 Dec 3;8(12):a030478. doi: 10.1101/cshperspect.a030478. Cold Spring Harb Perspect Med. 2018. PMID: 29530945 Free PMC article. Review.
-
Crosstalk of the Androgen Receptor with Transcriptional Collaborators: Potential Therapeutic Targets for Castration-Resistant Prostate Cancer.Cancers (Basel). 2017 Feb 28;9(3):22. doi: 10.3390/cancers9030022. Cancers (Basel). 2017. PMID: 28264478 Free PMC article. Review.
-
Androgen receptor splice variants and prostate cancer: From bench to bedside.Oncotarget. 2017 Mar 14;8(11):18550-18576. doi: 10.18632/oncotarget.14537. Oncotarget. 2017. PMID: 28077788 Free PMC article. Review.
References
-
- Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–7. discussion 7. - PubMed
-
- Labrie F, Dupont A, Belanger A, et al. New approach in the treatment of prostate cancer: complete instead of partial withdrawal of androgens. Prostate. 1983;4:579–94. - PubMed
-
- Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–71. - PubMed
-
- Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11:459–76. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30 DK092926/DK/NIDDK NIH HHS/United States
- P30 CA046592/CA/NCI NIH HHS/United States
- P50 CA69568/CA/NCI NIH HHS/United States
- R21 HD075005/HD/NICHD NIH HHS/United States
- R01 DK56356/DK/NIDDK NIH HHS/United States
- T32 GM007544/GM/NIGMS NIH HHS/United States
- P50 CA069568/CA/NCI NIH HHS/United States
- R01 DK056356/DK/NIDDK NIH HHS/United States
- T32 HD075005/HD/NICHD NIH HHS/United States
- 5 P30 CA46592/CA/NCI NIH HHS/United States
- T32 HD007505/HD/NICHD NIH HHS/United States
- P60 DK020572/DK/NIDDK NIH HHS/United States
- 5 P60 DK20572/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
