

Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus
- PMID: 19369296
- PMCID: PMC2694691
- DOI: 10.1093/hmg/ddp181
Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus
Abstract
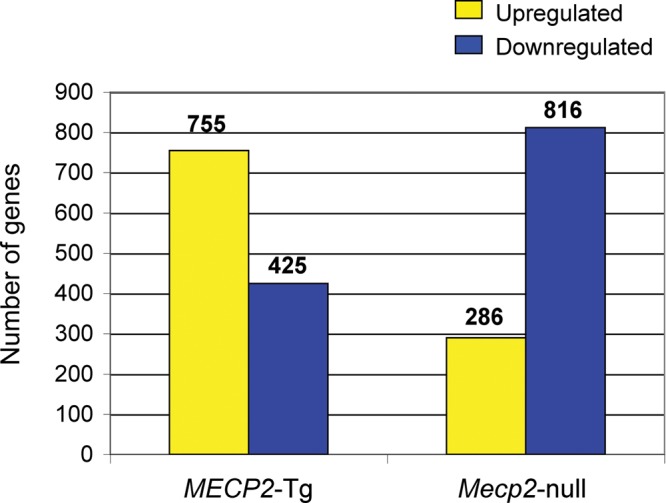
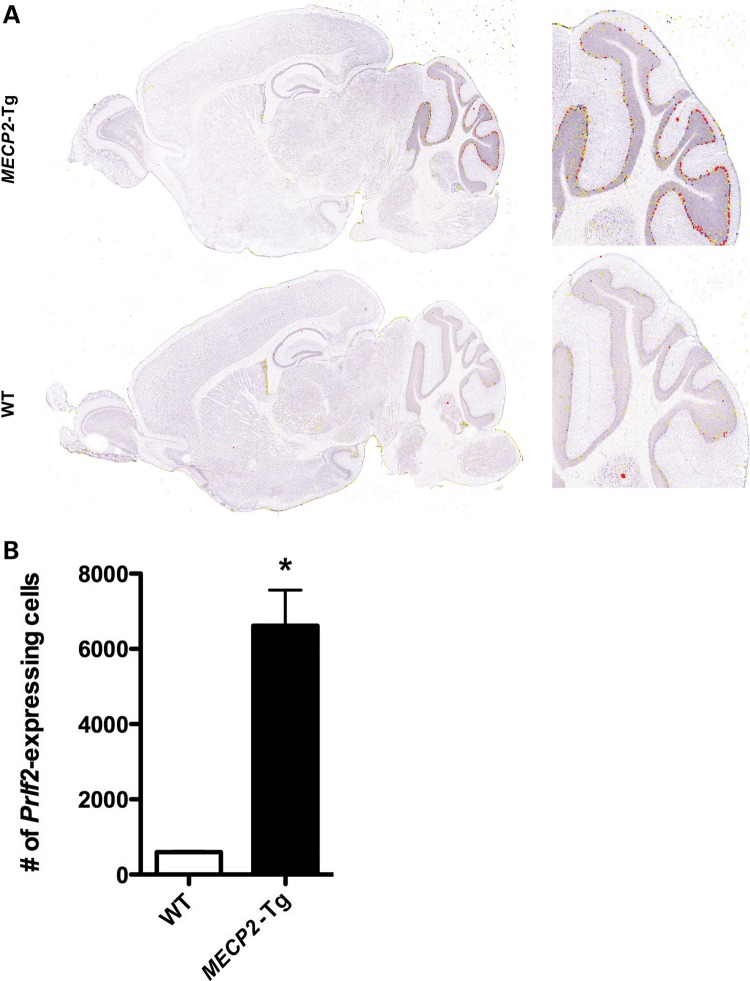
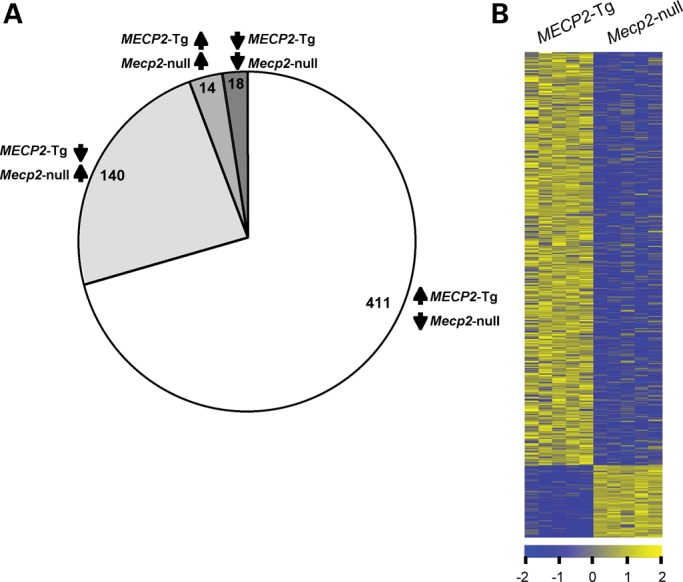
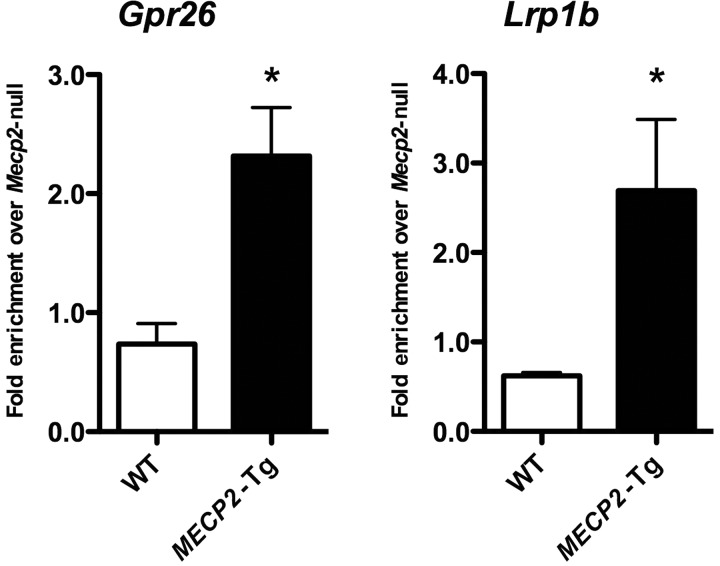
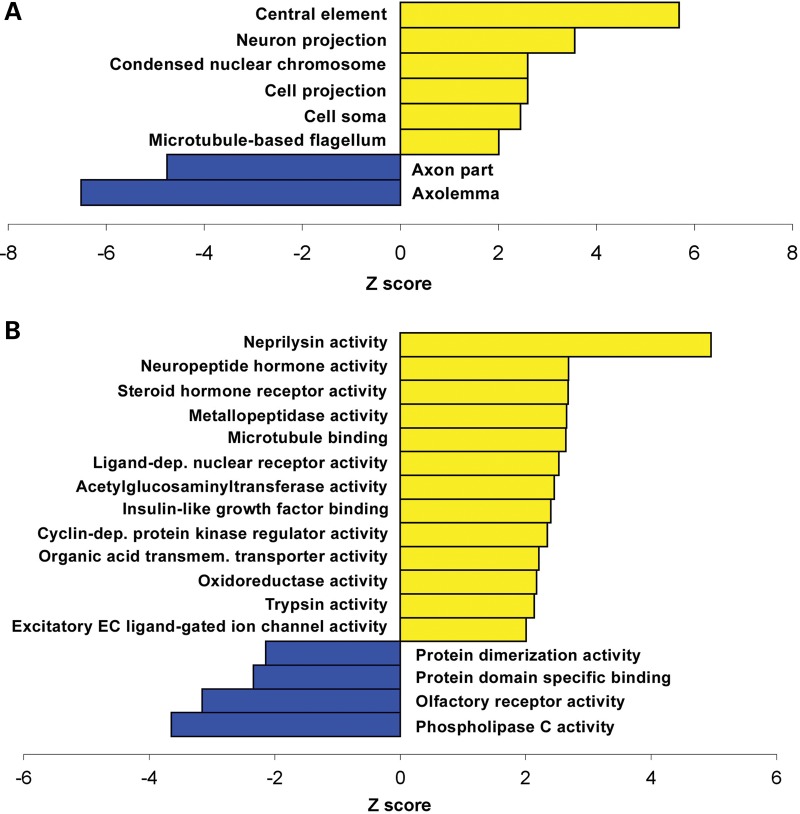
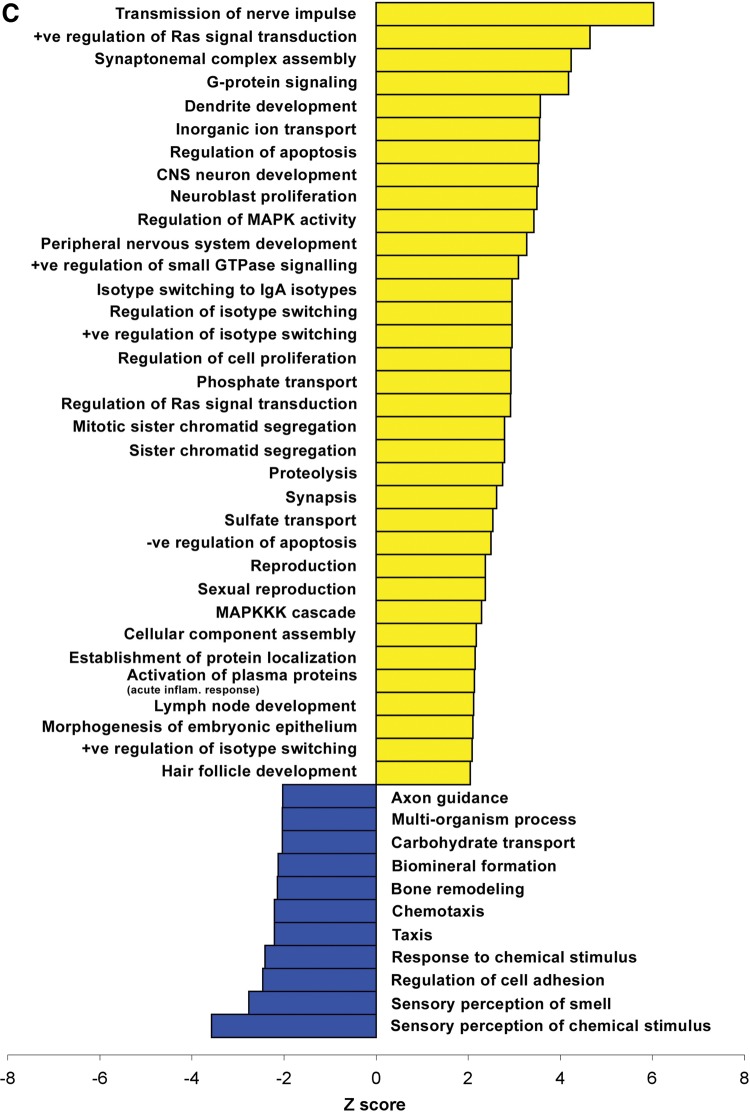
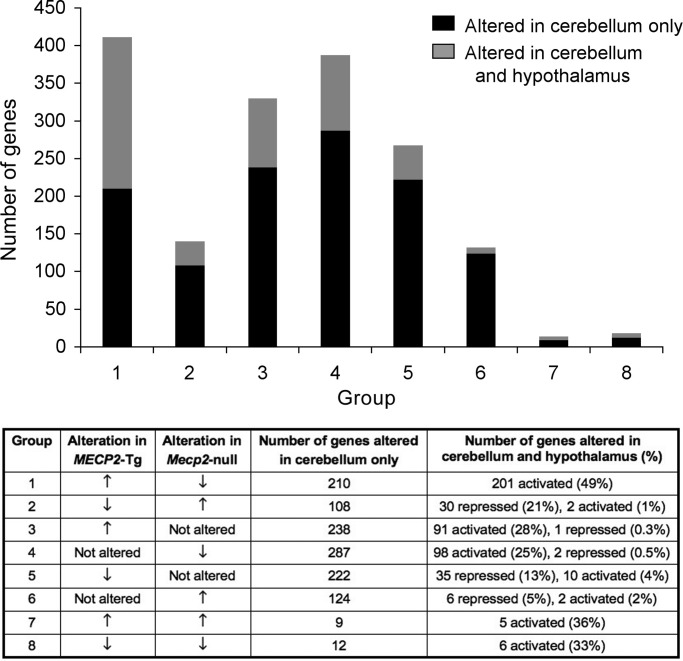
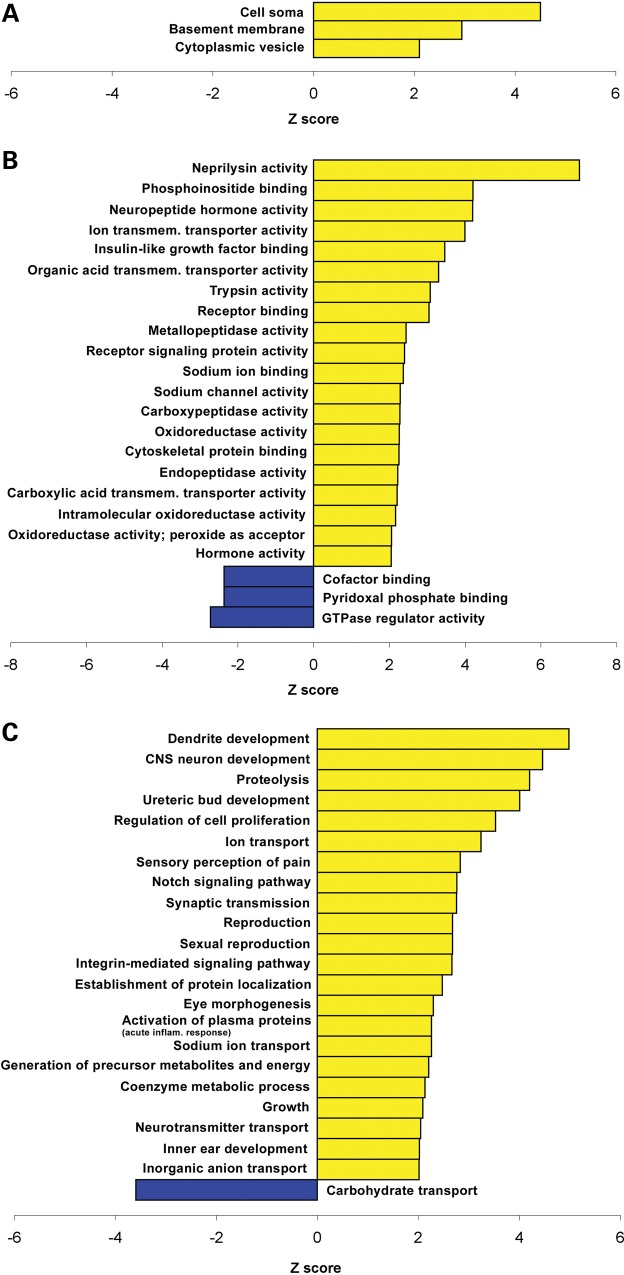
A group of post-natal neurodevelopmental disorders collectively referred to as MeCP2 disorders are caused by aberrations in the gene encoding methyl-CpG-binding protein 2 (MECP2). Loss of MeCP2 function causes Rett syndrome (RTT), whereas increased copy number of the gene causes MECP2 duplication or triplication syndromes. MeCP2 acts as a transcriptional repressor, however the gene expression changes observed in the hypothalamus of MeCP2 disorder mouse models suggest that MeCP2 can also upregulate gene expression, given that the majority of genes are downregulated upon loss of MeCP2 and upregulated in its presence. To determine if this dual role of MeCP2 extends beyond the hypothalamus, we studied gene expression patterns in the cerebellum of Mecp2-null and MECP2-Tg mice, modeling RTT and MECP2 duplication syndrome, respectively. We found that abnormal MeCP2 dosage causes alterations in the expression of hundreds of genes in the cerebellum. The majority of genes were upregulated in MECP2-Tg mice and downregulated in Mecp2-null mice, consistent with a role for MeCP2 as a modulator that can both increase and decrease gene expression. Interestingly, many of the genes altered in the cerebellum, particularly those increased by the presence of MeCP2 and decreased in its absence, were similarly altered in the hypothalamus. Our data suggest that either gain or loss of MeCP2 results in gene expression changes in multiple brain regions and that some of these changes are global. Further delineation of the expression pattern of MeCP2 target genes throughout the brain might identify subsets of genes that are more amenable to manipulation, and can thus be used to modulate some of the disease phenotypes.
Figures
References
-
- Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. - PubMed
-
- Chahrour M., Zoghbi H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. - PubMed
-
- Friez M.J., Jones J.R., Clarkson K., Lubs H., Abuelo D., Bier J.A., Pai S., Simensen R., Williams C., Giampietro P.F., et al. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118:e1687–e1695. - PubMed
-
- Lugtenberg D., de Brouwer A.P., Kleefstra T., Oudakker A.R., Frints S.G., Schrander-Stumpel C.T., Fryns J.P., Jensen L.R., Chelly J., Moraine C., et al. Chromosomal copy number changes in patients with non-syndromic X linked mental retardation detected by array CGH. J. Med. Genet. 2006;43:362–370. - PMC - PubMed
-
- Van Esch H., Bauters M., Ignatius J., Jansen M., Raynaud M., Hollanders K., Lugtenberg D., Bienvenu T., Jensen L.R., Gecz J., et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005;77:442–453. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
