

DOCK 6: combining techniques to model RNA-small molecule complexes
- PMID: 19369428
- PMCID: PMC2685511
- DOI: 10.1261/rna.1563609
DOCK 6: combining techniques to model RNA-small molecule complexes
Abstract
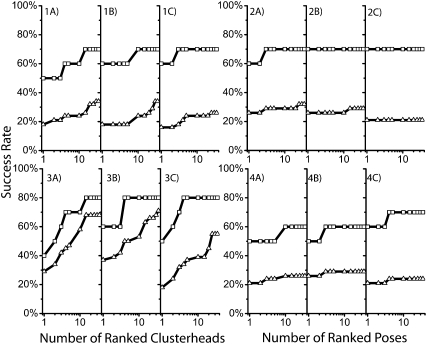
With an increasing interest in RNA therapeutics and for targeting RNA to treat disease, there is a need for the tools used in protein-based drug design, particularly DOCKing algorithms, to be extended or adapted for nucleic acids. Here, we have compiled a test set of RNA-ligand complexes to validate the ability of the DOCK suite of programs to successfully recreate experimentally determined binding poses. With the optimized parameters and a minimal scoring function, 70% of the test set with less than seven rotatable ligand bonds and 26% of the test set with less than 13 rotatable bonds can be successfully recreated within 2 A heavy-atom RMSD. When DOCKed conformations are rescored with the implicit solvent models AMBER generalized Born with solvent-accessible surface area (GB/SA) and Poisson-Boltzmann with solvent-accessible surface area (PB/SA) in combination with explicit water molecules and sodium counterions, the success rate increases to 80% with PB/SA for less than seven rotatable bonds and 58% with AMBER GB/SA and 47% with PB/SA for less than 13 rotatable bonds. These results indicate that DOCK can indeed be useful for structure-based drug design aimed at RNA. Our studies also suggest that RNA-directed ligands often differ from typical protein-ligand complexes in their electrostatic properties, but these differences can be accommodated through the choice of potential function. In addition, in the course of the study, we explore a variety of newly added DOCK functions, demonstrating the ease with which new functions can be added to address new scientific questions.
Figures





References
-
- Bannwarth S., Gatignol A. HIV-1 TAR RNA: The target of molecular interactions between the virus and its host. Curr. HIV Res. 2005;3:61–71. - PubMed
-
- Bayly C.I., Cieplak P., Cornell W.D., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993;97:10269–10280.
-
- Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources