

Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome
- PMID: 19372089
- PMCID: PMC3395372
- DOI: 10.1136/jmg.2008.063412
Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome
Abstract
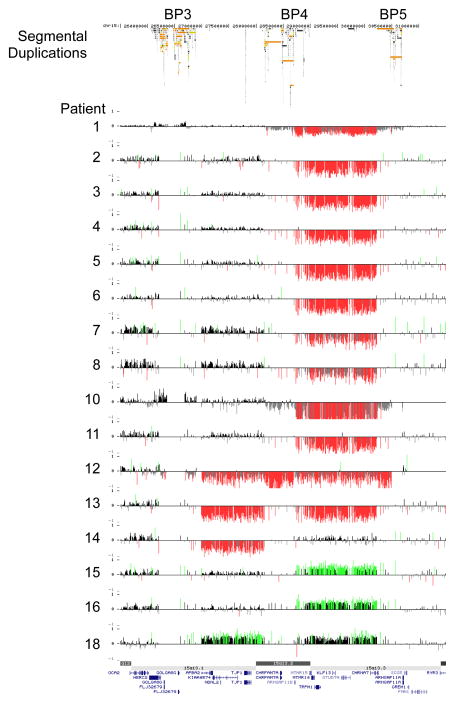
Background: Recurrent 15q13.3 microdeletions were recently identified with identical proximal (BP4) and distal (BP5) breakpoints and associated with mild to moderate mental retardation and epilepsy.
Methods: To assess further the clinical implications of this novel 15q13.3 microdeletion syndrome, 18 new probands with a deletion were molecularly and clinically characterised. In addition, we evaluated the characteristics of a family with a more proximal deletion between BP3 and BP4. Finally, four patients with a duplication in the BP3-BP4-BP5 region were included in this study to ascertain the clinical significance of duplications in this region.
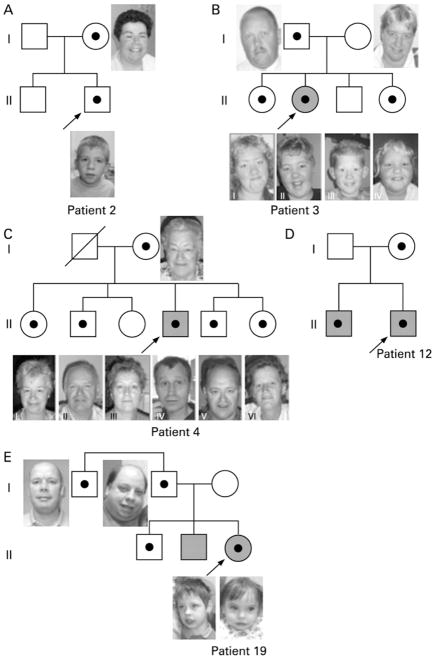
Results: The 15q13.3 microdeletion in our series was associated with a highly variable intra- and inter-familial phenotype. At least 11 of the 18 deletions identified were inherited. Moreover, 7 of 10 siblings from four different families also had this deletion: one had a mild developmental delay, four had only learning problems during childhood, but functioned well in daily life as adults, whereas the other two had no learning problems at all. In contrast to previous findings, seizures were not a common feature in our series (only 2 of 17 living probands). Three patients with deletions had cardiac defects and deletion of the KLF13 gene, located in the critical region, may contribute to these abnormalities. The limited data from the single family with the more proximal BP3-BP4 deletion suggest this deletion may have little clinical significance. Patients with duplications of the BP3-BP4-BP5 region did not share a recognisable phenotype, but psychiatric disease was noted in 2 of 4 patients.
Conclusions: Overall, our findings broaden the phenotypic spectrum associated with 15q13.3 deletions and suggest that, in some individuals, deletion of 15q13.3 is not sufficient to cause disease. The existence of microdeletion syndromes, associated with an unpredictable and variable phenotypic outcome, will pose the clinician with diagnostic difficulties and challenge the commonly used paradigm in the diagnostic setting that aberrations inherited from a phenotypically normal parent are usually without clinical consequences.
Figures

Comment in
-
"New microdeletion syndromes: complex, but no new paradigms".J Med Genet. 2009 Aug;46(8):576. doi: 10.1136/jmg.2009.068916. J Med Genet. 2009. PMID: 19648125 No abstract available.
References
-
- Koolen DA, Vissers LE, Pfundt R, de LN, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, Knoers NV, van Kessel AG, Sistermans EA, Veltman JA, Brunner HG, de Vries BB. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006 Sep;38(9):999–1001. - PubMed
-
- Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, Striano P, Coppola A, Regan R, Price SM, Knoers NV, Eis PS, Brunner HG, Hennekam RC, Knight SJ, de Vries BB, Zuffardi O, Eichler EE. Characterization of a recurrent 15q24 microdeletion syndrome. Hum Mol Genet. 2007 Mar 1;16(5):567–72. - PubMed
-
- Willatt L, Cox J, Barber J, Cabanas ED, Collins A, Donnai D, FitzPatrick DR, Maher E, Martin H, Parnau J, Pindar L, Ramsay J, Shaw-Smith C, Sistermans EA, Tettenborn M, Trump D, de Vries BB, Walker K, Raymond FL. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005 Jul;77(1):154–60. - PMC - PubMed
-
- Ensenauer RE, Adeyinka A, Flynn HC, Michels VV, Lindor NM, Dawson DB, Thorland EC, Lorentz CP, Goldstein JL, McDonald MT, Smith WE, Simon-Fayard E, Alexander AA, Kulharya AS, Ketterling RP, Clark RD, Jalal SM. Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am J Hum Genet. 2003 Nov;73(5):1027–40. - PMC - PubMed
-
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998 Oct;14(10):417–22. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases