

Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2
- PMID: 19372218
- PMCID: PMC2713561
- DOI: 10.1074/jbc.M808262200
Dynamic partnership between KCNQ1 and KCNE1 and influence on cardiac IKs current amplitude by KCNE2
Abstract
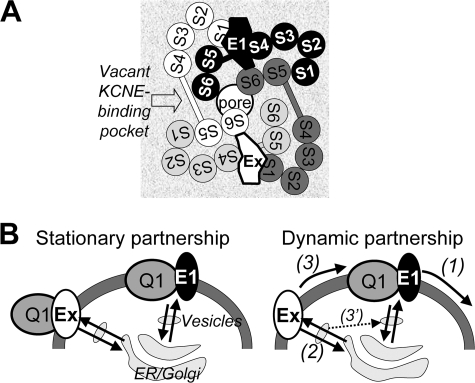
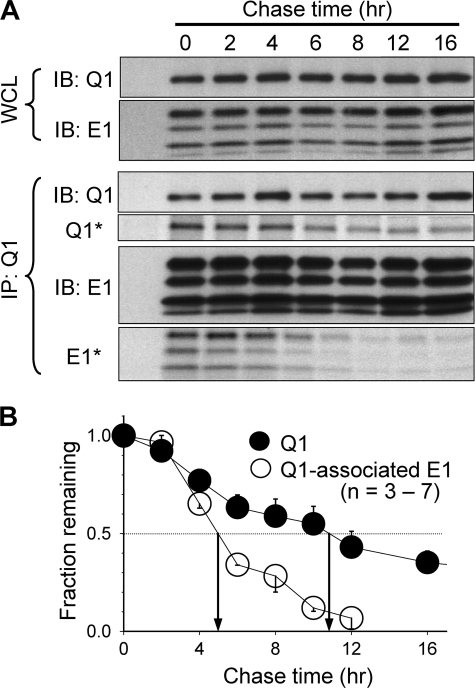
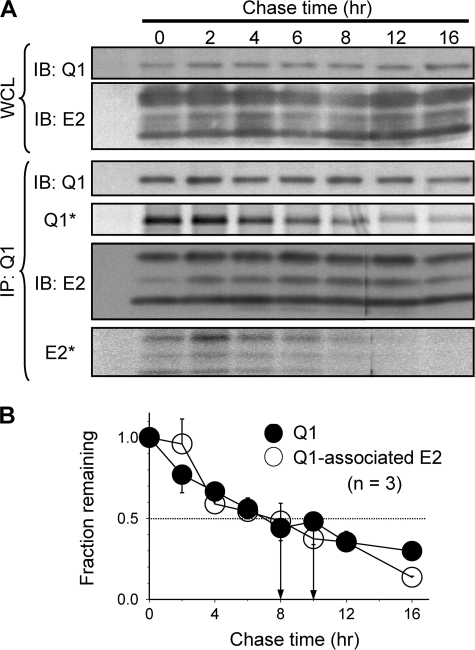
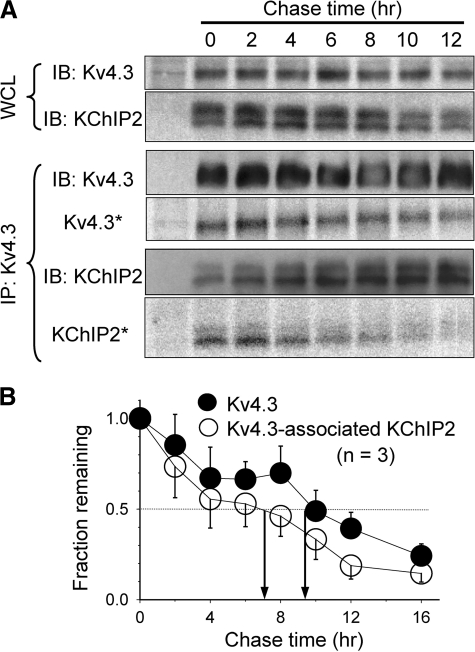
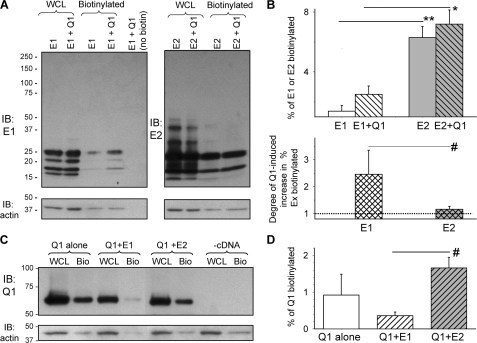
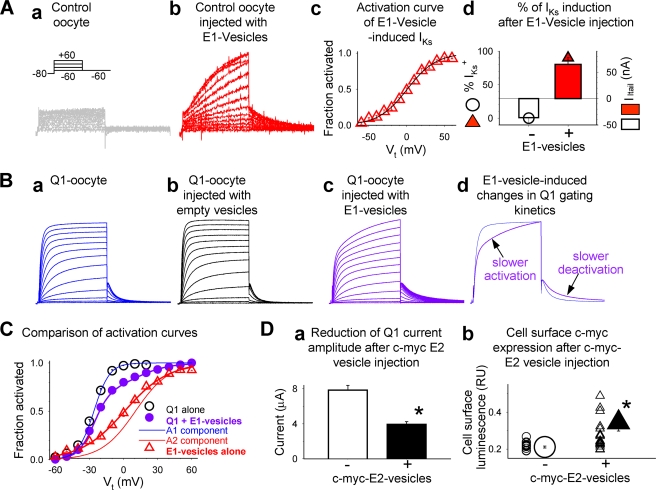
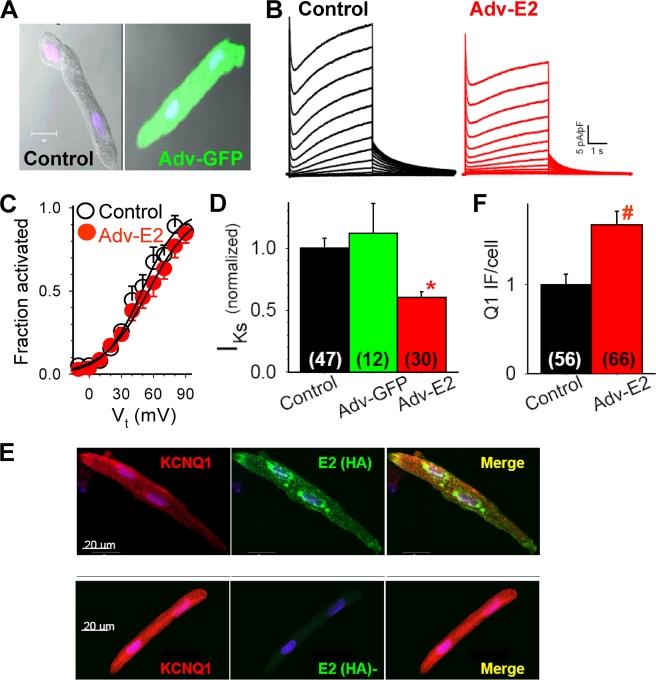
Cardiac slow delayed rectifier (IKs) channel is composed of KCNQ1 (pore-forming) and KCNE1 (auxiliary) subunits. Although KCNE1 is an obligate IKs component that confers the uniquely slow gating kinetics, KCNE2 is also expressed in human heart. In vitro experiments suggest that KCNE2 can associate with the KCNQ1-KCNE1 complex to suppress the current amplitude without altering the slow gating kinetics. Our goal here is to test the role of KCNE2 in cardiac IKs channel function. Pulse-chase experiments in COS-7 cells show that there is a KCNE1 turnover in the KCNQ1-KCNE1 complex, supporting the possibility that KCNE1 in the IKs channel complex can be substituted by KCNE2 when the latter is available. Biotinylation experiments in COS-7 cells show that although KCNE1 relies on KCNQ1 coassembly for more efficient cell surface expression, KCNE2 can independently traffic to the cell surface, thus becoming available for substituting KCNE1 in the IKs channel complex. Injecting vesicles carrying KCNE1 or KCNE2 into KCNQ1-expressing oocytes leads to KCNQ1 modulation in the same manner as KCNQ1+KCNEx (where x=1 or 2) cRNA coinjection. Thus, free KCNEx peptides delivered to the cell membrane can associate with existing KCNQ1 channels to modulate their function. Finally, adenovirus-mediated KCNE2 expression in adult guinea pig ventricular myocytes exhibited colocalization with native KCNQ1 protein and reduces the native IKs current density. We propose that in cardiac myocytes the IKs current amplitude is under dynamic control by the availability of KCNE2 subunits in the cell membrane.
Figures
Similar articles
-
Structural basis for K(V)7.1-KCNE(x) interactions in the I(Ks) channel complex.Heart Rhythm. 2010 May;7(5):708-13. doi: 10.1016/j.hrthm.2009.12.017. Epub 2009 Dec 24. Heart Rhythm. 2010. PMID: 20206317 Free PMC article. Review.
-
Adult Ventricular Myocytes Segregate KCNQ1 and KCNE1 to Keep the IKs Amplitude in Check Until When Larger IKs Is Needed.Circ Arrhythm Electrophysiol. 2017 Jun;10(6):e005084. doi: 10.1161/CIRCEP.117.005084. Circ Arrhythm Electrophysiol. 2017. PMID: 28611207 Free PMC article.
-
KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of I(Ks) current amplitude in the heart.Heart Rhythm. 2006 Dec;3(12):1469-80. doi: 10.1016/j.hrthm.2006.08.019. Epub 2006 Aug 25. Heart Rhythm. 2006. PMID: 17161791
-
Probing the structural basis for differential KCNQ1 modulation by KCNE1 and KCNE2.J Gen Physiol. 2012 Dec;140(6):653-69. doi: 10.1085/jgp.201210847. J Gen Physiol. 2012. PMID: 23183700 Free PMC article.
-
Insights into Cardiac IKs (KCNQ1/KCNE1) Channels Regulation.Int J Mol Sci. 2020 Dec 11;21(24):9440. doi: 10.3390/ijms21249440. Int J Mol Sci. 2020. PMID: 33322401 Free PMC article. Review.
Cited by
-
A Possible Explanation for the Low Penetrance of Pathogenic KCNE1 Variants in Long QT Syndrome Type 5.Pharmaceuticals (Basel). 2022 Dec 13;15(12):1550. doi: 10.3390/ph15121550. Pharmaceuticals (Basel). 2022. PMID: 36559002 Free PMC article.
-
Acute Adenoviral Infection Elicits an Arrhythmogenic Substrate Prior to Myocarditis.Circ Res. 2024 Mar 29;134(7):892-912. doi: 10.1161/CIRCRESAHA.122.322437. Epub 2024 Feb 28. Circ Res. 2024. PMID: 38415360 Free PMC article.
-
Structural basis for K(V)7.1-KCNE(x) interactions in the I(Ks) channel complex.Heart Rhythm. 2010 May;7(5):708-13. doi: 10.1016/j.hrthm.2009.12.017. Epub 2009 Dec 24. Heart Rhythm. 2010. PMID: 20206317 Free PMC article. Review.
-
Adult Ventricular Myocytes Segregate KCNQ1 and KCNE1 to Keep the IKs Amplitude in Check Until When Larger IKs Is Needed.Circ Arrhythm Electrophysiol. 2017 Jun;10(6):e005084. doi: 10.1161/CIRCEP.117.005084. Circ Arrhythm Electrophysiol. 2017. PMID: 28611207 Free PMC article.
-
KCNE2 protein is more abundant in ventricles than in atria and can accelerate hERG protein degradation in a phosphorylation-dependent manner.Am J Physiol Heart Circ Physiol. 2012 Feb 15;302(4):H910-22. doi: 10.1152/ajpheart.00691.2011. Epub 2011 Dec 16. Am J Physiol Heart Circ Physiol. 2012. PMID: 22180649 Free PMC article.
References
-
- Jost M., Virág L., Bitay M., Takács J., Lengyel C., Biliczki P., Nagy Z., Bogáts G., Lathrop D. A., Papp J. G., Varró A. ( 2005) Circulation 112, 1392– 1399 - PubMed
-
- Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. ( 1996) Nature 384, 78– 80 - PubMed
-
- Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., Keating M. T. ( 1996) Nature 384, 80– 83 - PubMed
-
- Chen H., Kim L. A., Rajan S., Xu S., Goldstein S. A. N. ( 2003) Neuron 40, 15– 23 - PubMed
-
- Tapper A. R., George A. L., Jr. ( 2001) J. Biol. Chem. 276, 38249– 38254 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
