

DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy
- PMID: 19372220
- PMCID: PMC2719367
- DOI: 10.1074/jbc.M109.006759
DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy
Abstract
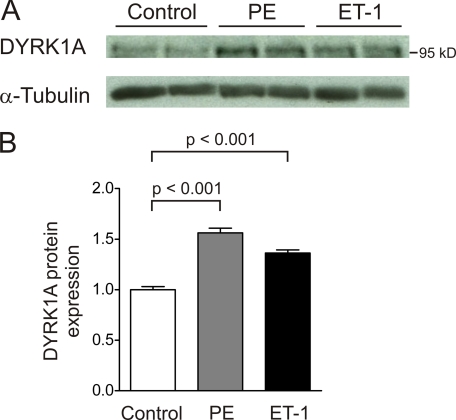
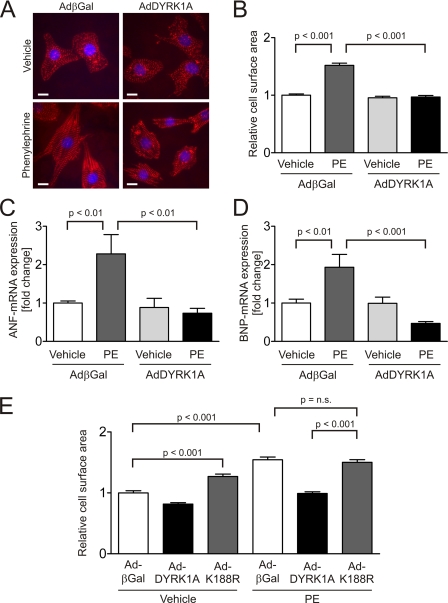
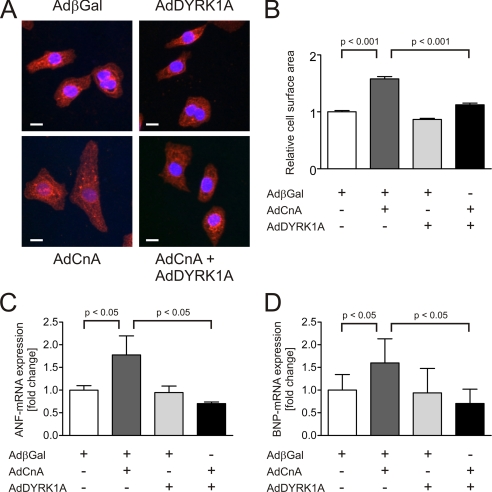
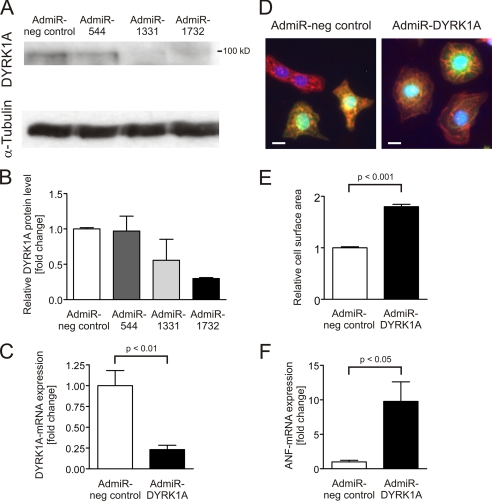
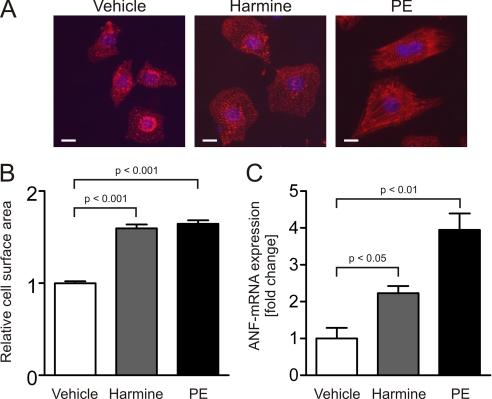
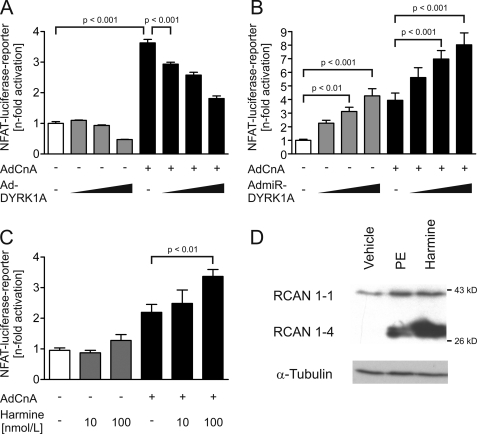
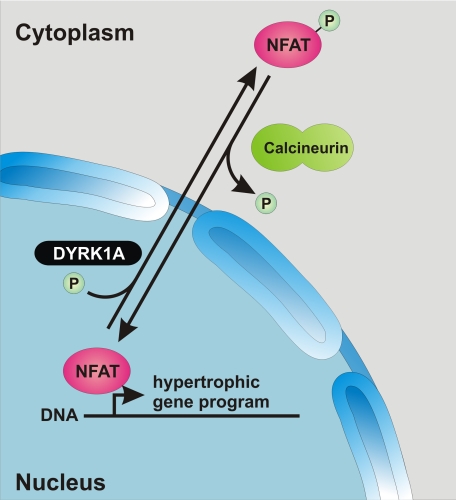
Activation of the phosphatase calcineurin and its downstream targets, transcription factors of the NFAT family, results in cardiomyocyte hypertrophy. Recently, it has been shown that the dual specificity tyrosine (Y) phosphorylation-regulated kinase 1A (DYRK1A) is able to antagonize calcineurin signaling by directly phosphorylating NFATs. We thus hypothesized that DYRK1A might modulate the hypertrophic response of cardiomyocytes. In a model of phenylephrine-induced hypertrophy, adenovirus-mediated overexpression of DYKR1A completely abrogated the hypertrophic response and significantly reduced the expression of the natriuretic peptides ANF and BNP. Furthermore, DYRK1A blunted cardiomyocyte hypertrophy induced by overexpression of constitutively active calcineurin and attenuated the induction of the hypertrophic gene program. Conversely, knockdown of DYRK1A, utilizing adenoviruses encoding for a specific synthetic miRNA, resulted in an increase in cell surface area accompanied by up-regulation of ANF- mRNA. Similarly, treatment of cardiomyocytes with harmine, a specific inhibitor of DYRK1A, revealed cardiomyocyte hypertrophy on morphological and molecular level. Moreover, constitutively active calcineurin led to robust induction of an NFAT-dependent luciferase reporter, whereas DYRK1A attenuated calcineurin-induced reporter activation in cardiomyocytes. Conversely, both knockdown and pharmacological inhibition of DYRK1A significantly augmented the effect of calcineurin in this assay. In summary, we identified DYRK1A as a novel negative regulator of cardiomyocyte hypertrophy. Mechanistically, this effect appears to be mediated via inhibition of NFAT transcription factors.
Figures
Similar articles
-
Enhanced expression of DYRK1A in cardiomyocytes inhibits acute NFAT activation but does not prevent hypertrophy in vivo.Cardiovasc Res. 2011 Jun 1;90(3):521-8. doi: 10.1093/cvr/cvr023. Epub 2011 Jan 27. Cardiovasc Res. 2011. PMID: 21273244
-
Dyrk1a regulates the cardiomyocyte cell cycle via D-cyclin-dependent Rb/E2f-signalling.Cardiovasc Res. 2016 Jun 1;110(3):381-94. doi: 10.1093/cvr/cvw074. Epub 2016 Apr 7. Cardiovasc Res. 2016. PMID: 27056896
-
PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling.Circ Res. 2008 Mar 28;102(6):711-9. doi: 10.1161/CIRCRESAHA.107.165985. Epub 2008 Feb 7. Circ Res. 2008. PMID: 18258855
-
Calcium-calcineurin signaling in the regulation of cardiac hypertrophy.Biochem Biophys Res Commun. 2004 Oct 1;322(4):1178-91. doi: 10.1016/j.bbrc.2004.07.121. Biochem Biophys Res Commun. 2004. PMID: 15336966 Review.
-
DYRK1A inhibitors for disease therapy: Current status and perspectives.Eur J Med Chem. 2022 Feb 5;229:114062. doi: 10.1016/j.ejmech.2021.114062. Epub 2021 Dec 21. Eur J Med Chem. 2022. PMID: 34954592 Review.
Cited by
-
Dyrk1A, a serine/threonine kinase, is involved in ERK and Akt activation in the brain of hyperhomocysteinemic mice.Mol Neurobiol. 2013 Feb;47(1):105-16. doi: 10.1007/s12035-012-8326-1. Epub 2012 Aug 26. Mol Neurobiol. 2013. PMID: 22923366
-
Salivary cardiac-enriched FHL2-interacting protein is associated with higher diastolic-to-systolic-blood pressure ratio, sedentary time and center of pressure displacement in healthy 7-9 years old school-children.Front Endocrinol (Lausanne). 2024 Jan 18;15:1292653. doi: 10.3389/fendo.2024.1292653. eCollection 2024. Front Endocrinol (Lausanne). 2024. PMID: 38304464 Free PMC article.
-
Primers on molecular pathways--the NFAT transcription pathway in pancreatic cancer.Pancreatology. 2010;10(4):416-22. doi: 10.1159/000315035. Epub 2010 Aug 19. Pancreatology. 2010. PMID: 20720442 Free PMC article. Review.
-
Liquid-Liquid Phase Separation in Cardiovascular Diseases.Cells. 2022 Sep 28;11(19):3040. doi: 10.3390/cells11193040. Cells. 2022. PMID: 36231002 Free PMC article. Review.
-
A Bioinformatics Evaluation of the Role of Dual-Specificity Tyrosine-Regulated Kinases in Colorectal Cancer.Cancers (Basel). 2022 Apr 18;14(8):2034. doi: 10.3390/cancers14082034. Cancers (Basel). 2022. PMID: 35454940 Free PMC article.
References
-
- Levy D., Garrison R. J., Savage D. D., Kannel W. B., Castelli W. P. ( 1990) N. Engl. J. Med. 322, 1561– 1566 - PubMed
-
- Frey N., Olson E. N. ( 2003) Annu. Rev. Physiol. 65, 45– 79 - PubMed
-
- Hill J. A., Olson E. N. ( 2008) N. Engl. J. Med. 358, 1370– 1380 - PubMed
-
- Frey N., Katus H. A., Olson E. N., Hill J. A. ( 2004) Circulation 109, 1580– 1589 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
