

KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer
- PMID: 19372556
- PMCID: PMC2729756
- DOI: 10.1158/1535-7163.MCT-08-0972
KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer
Abstract
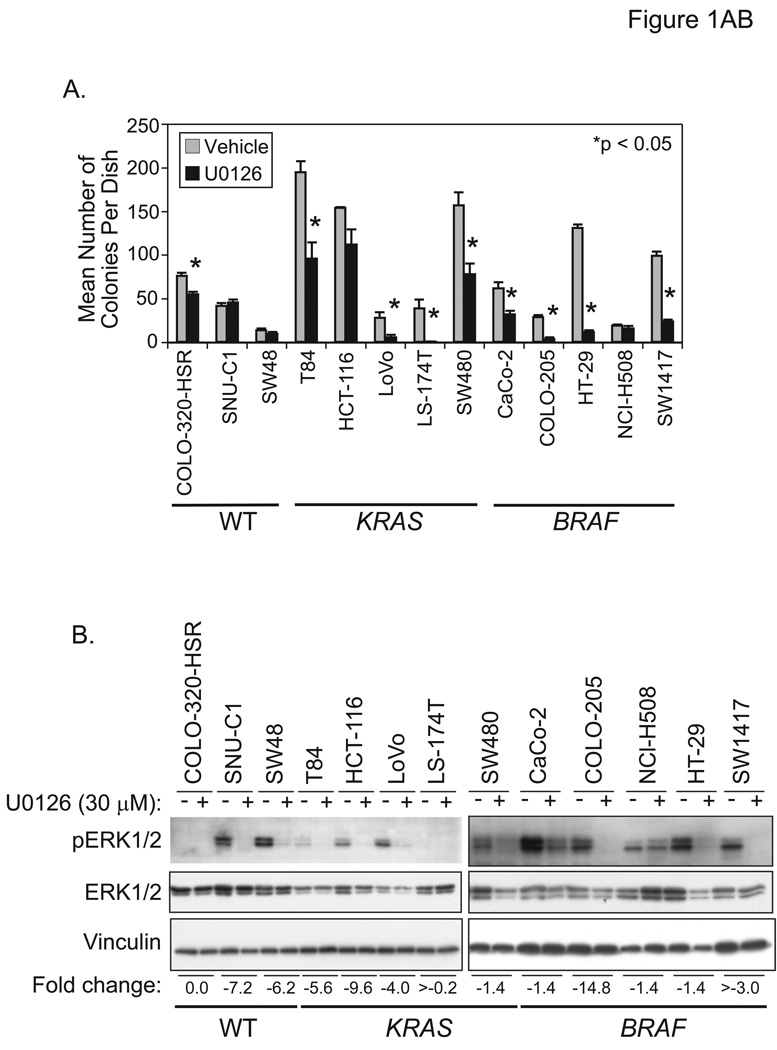
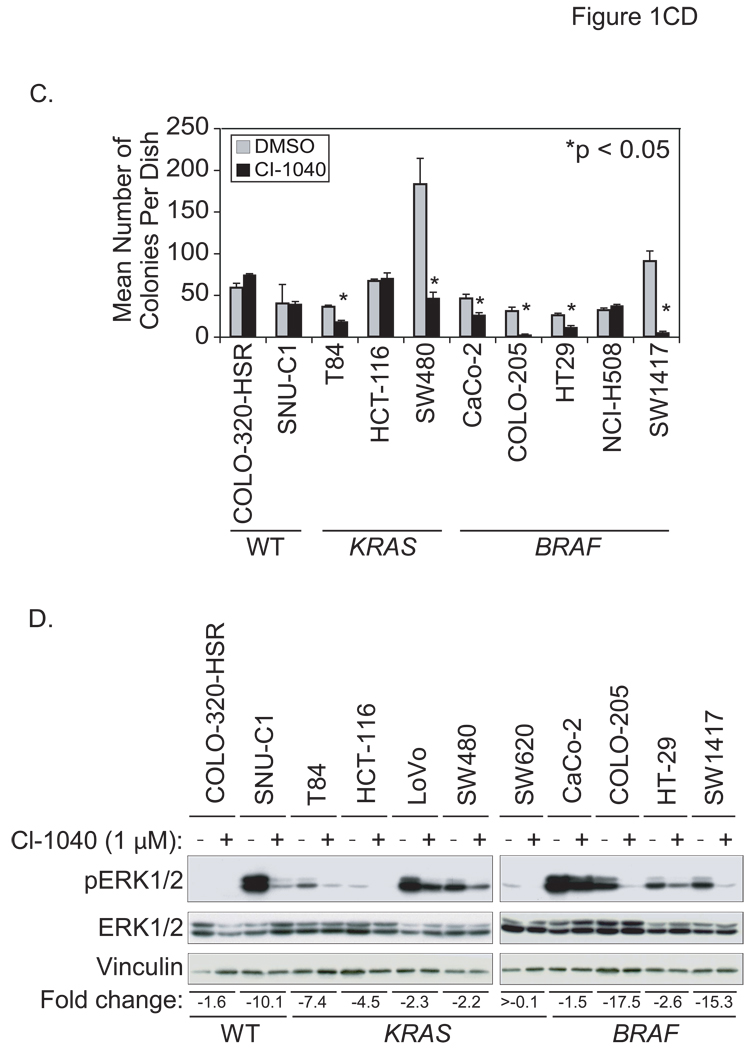
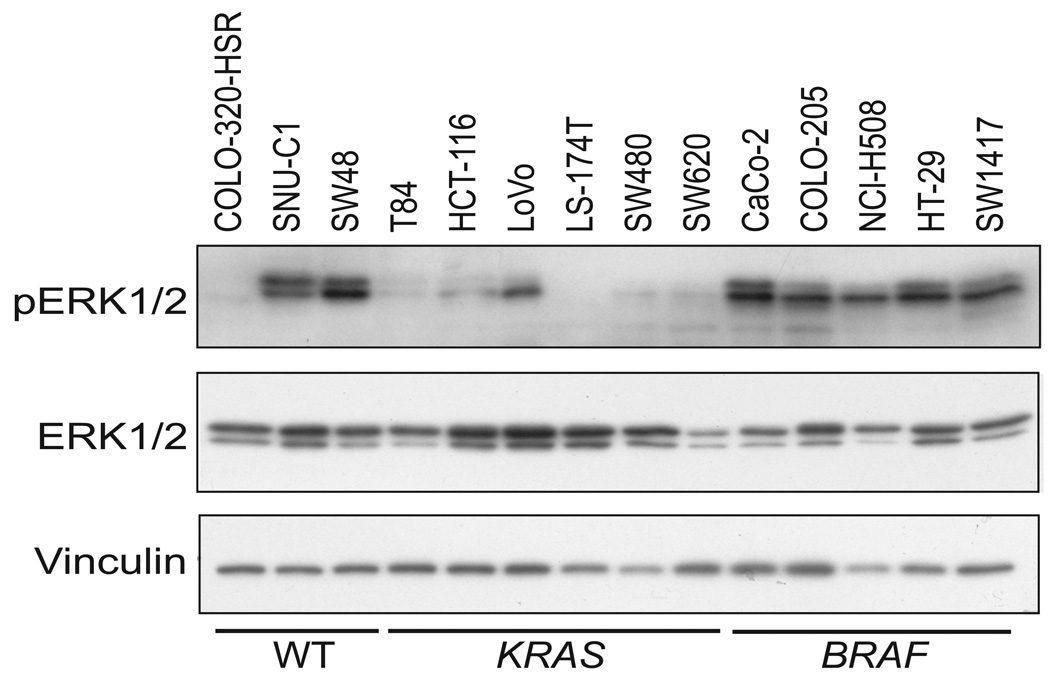
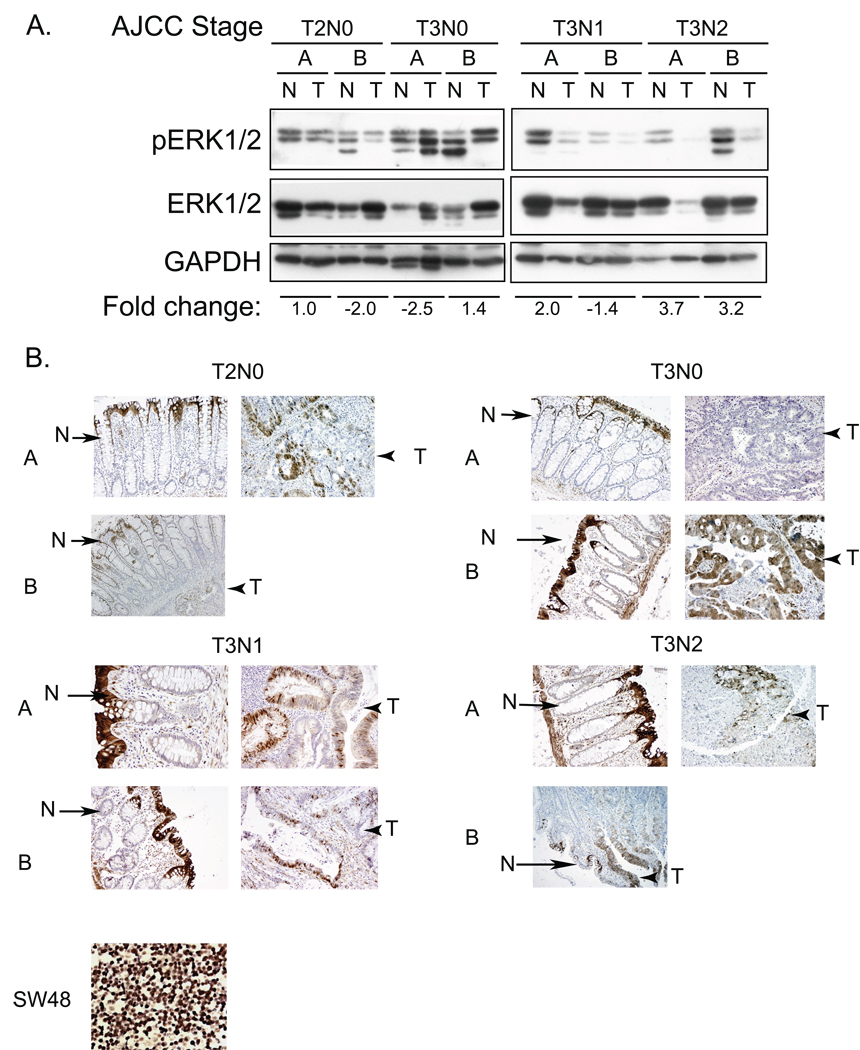
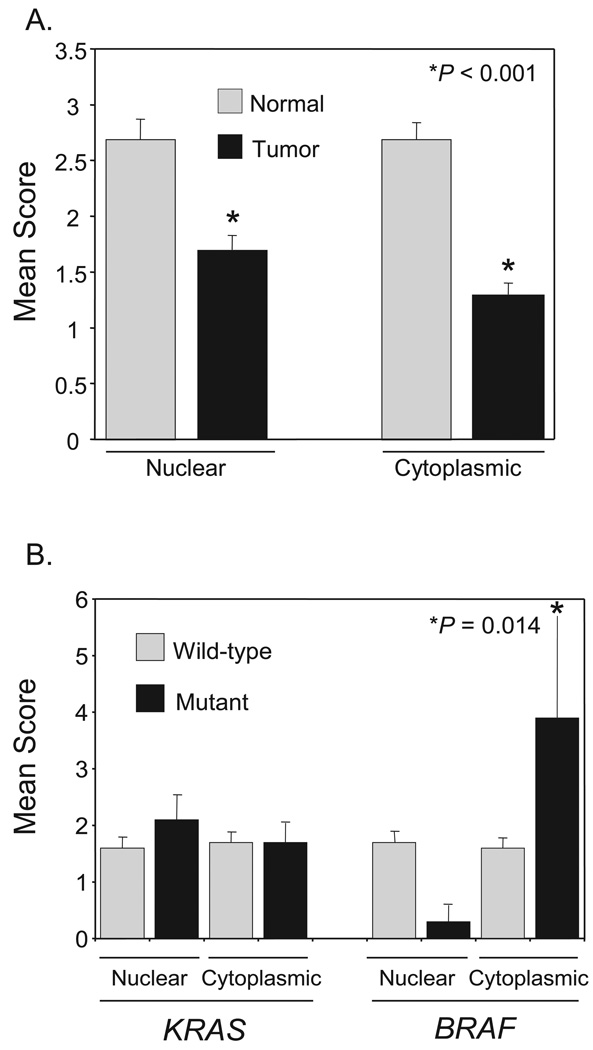
Phase II clinical trials of mitogen-activated protein/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitors are ongoing and ERK1/2 activation is frequently used as a biomarker. In light of the mutational activation of BRAF and KRAS in colorectal cancer, inhibitors of the Raf-MEK-ERK mitogen-activated protein kinase are anticipated to be promising. Previous studies in pancreatic cancer have found little correlation between BRAF/KRAS mutation status and ERK1/2 activation, suggesting that identifying biomarkers of MEK inhibitor response may be more challenging than previously thought. The purpose of this study was to evaluate the effectiveness of MEK inhibitor therapy for colorectal cancer and BRAF/KRAS mutation status and ERK1/2 activation as biomarkers for MEK inhibitor therapy. First, we found that MEK inhibitor treatment impaired the anchorage-independent growth of nearly all KRAS/BRAF mutant, but not wild-type, colorectal cancer cells. There was a correlation between BRAF, but not KRAS, mutation status and ERK1/2 activation. Second, neither elevated ERK1/2 activation nor reduction of ERK1/2 activity correlated with MEK inhibition of anchorage-independent growth. Finally, we validated our cell line observations and found that ERK1/2 activation correlated with BRAF, but not KRAS, mutation status in 190 patient colorectal cancer tissues. Surprisingly, we also found that ERK activation was elevated in normal colonic epithelium, suggesting that normal cell toxicity may be a complication for colorectal cancer treatment. Our results suggest that although MEK inhibitors show promise in colorectal cancer, KRAS/BRAF mutation status, but not ERK activation as previously thought, may be useful biomarkers for MEK inhibitor sensitivity.
Conflict of interest statement
Figures
Similar articles
-
Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines.Int J Cancer. 2009 Nov 15;125(10):2332-41. doi: 10.1002/ijc.24604. Int J Cancer. 2009. PMID: 19637312
-
Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells.Sci Signal. 2011 Mar 29;4(166):ra17. doi: 10.1126/scisignal.2001752. Sci Signal. 2011. Corrected and republished in: Sci Signal. 2011;4(170):er2. doi: 10.1126/scisignal.4170er2. PMID: 21447798 Corrected and republished.
-
KRAS or BRAF mutation status is a useful predictor of sensitivity to MEK inhibition in ovarian cancer.Br J Cancer. 2008 Dec 16;99(12):2020-8. doi: 10.1038/sj.bjc.6604783. Epub 2008 Nov 18. Br J Cancer. 2008. PMID: 19018267 Free PMC article.
-
Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy.Oncogene. 2016 May 19;35(20):2547-61. doi: 10.1038/onc.2015.329. Epub 2015 Sep 14. Oncogene. 2016. PMID: 26364606 Review.
-
Dual Inhibition of MEK and PI3K Pathway in KRAS and BRAF Mutated Colorectal Cancers.Int J Mol Sci. 2015 Sep 23;16(9):22976-88. doi: 10.3390/ijms160922976. Int J Mol Sci. 2015. PMID: 26404261 Free PMC article. Review.
Cited by
-
Reduced phosphorylation of ribosomal protein S6 is associated with sensitivity to MEK inhibition in gastric cancer cells.Cancer Sci. 2016 Dec;107(12):1919-1928. doi: 10.1111/cas.13094. Epub 2016 Dec 19. Cancer Sci. 2016. PMID: 27699948 Free PMC article.
-
Pharmacogenomic landscape of patient-derived tumor cells informs precision oncology therapy.Nat Genet. 2018 Oct;50(10):1399-1411. doi: 10.1038/s41588-018-0209-6. Epub 2018 Sep 27. Nat Genet. 2018. PMID: 30262818 Free PMC article.
-
Advances in the preclinical testing of cancer therapeutic hypotheses.Nat Rev Drug Discov. 2011 Mar;10(3):179-87. doi: 10.1038/nrd3385. Nat Rev Drug Discov. 2011. PMID: 21358737 Review.
-
RAS specific protease induces irreversible growth arrest via p27 in several KRAS mutant colorectal cancer cell lines.Sci Rep. 2021 Sep 9;11(1):17925. doi: 10.1038/s41598-021-97422-0. Sci Rep. 2021. PMID: 34504197 Free PMC article.
-
Oncogenic mutant RAS signaling activity is rescaled by the ERK/MAPK pathway.Mol Syst Biol. 2020 Oct;16(10):e9518. doi: 10.15252/msb.20209518. Mol Syst Biol. 2020. PMID: 33073539 Free PMC article.
References
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. - PubMed
-
- Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. - PubMed
-
- Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science (New York NY. 2006;314:268–274. - PubMed
-
- Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science (New York, NY. 2007;318:1108–1113. - PubMed
-
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science (New York, NY. 1993;260:85–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
