

NAD and axon degeneration: from the Wlds gene to neurochemistry
- PMID: 19372760
- PMCID: PMC2675153
- DOI: 10.4161/cam.3.1.7483
NAD and axon degeneration: from the Wlds gene to neurochemistry
Abstract
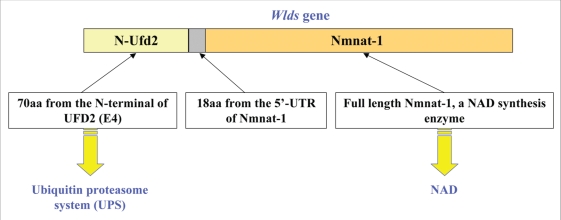
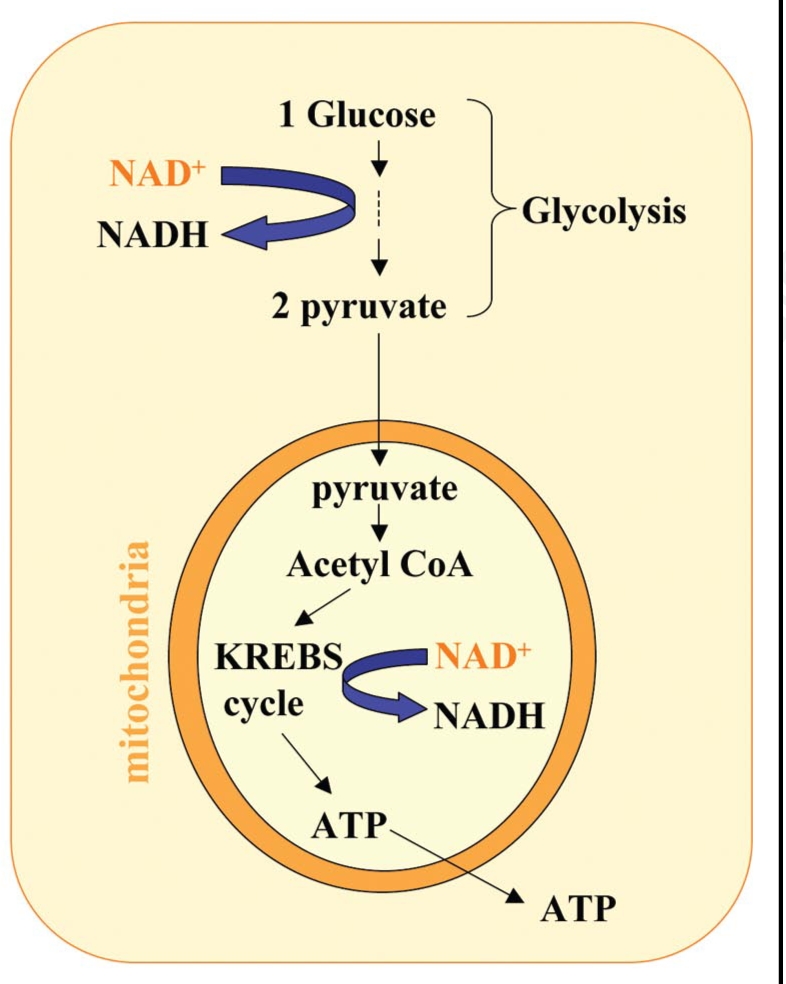
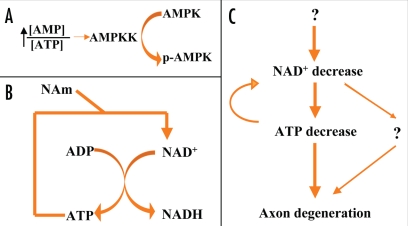
Neurodegenerative diseases have become a global issue due to the aging population. These disorders affect a vast patient population and represent a huge area of unmet therapeutic need. Axon degeneration is a common pathological character of those neurodegenerative diseases. It results in the loss of communication between neurons. Two decades ago, the Wallerian degeneration slow (Wlds) mouse strain was identified, in which the degeneration of transected axons is delayed. The phenotype is attributed to the overexpression of a chimeric protein Wlds which contains a short fragment of the ubiquitin assembly protein UFD2 and the full-length nicotinamide adenine dinucleotide (NAD) synthetic enzyme Nicotinamide mononucleotide adenylyl-transferase-1 (Nmnat-1). However, the underlying molecular mechanism remains largely unknown. Recently, it's reported by independent researchers that the full length coding sequence of mouse Nmnat-1 could mimic the axonal protective effect of the Wlds gene when overexpressed in primary neural cultures. Together with a significant number of subsequential reports, this finding highlighted the substantial role of nicotinamide adenine dinucleotide (NAD) in the process of axon degeneration. Here we reviewed the history of axon degeneration research from a neurochemical standpoint and discuss the potential involvement of NAD synthesis, NAD consumption and NAD-dependent proteins and small molecules in axon degeneration.
Figures

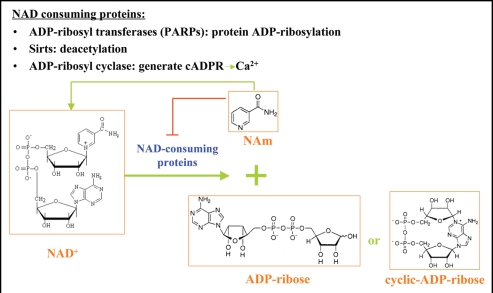

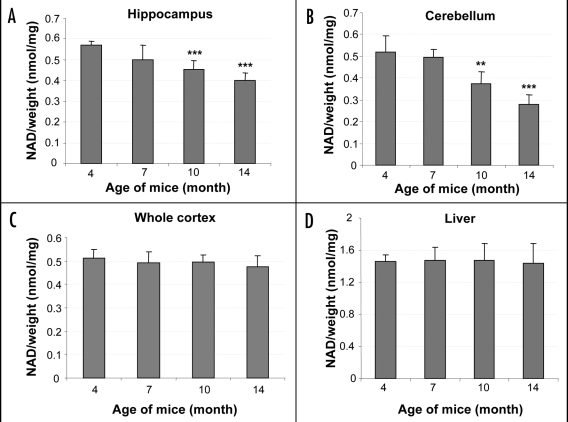
Similar articles
-
A local mechanism mediates NAD-dependent protection of axon degeneration.J Cell Biol. 2005 Aug 1;170(3):349-55. doi: 10.1083/jcb.200504028. Epub 2005 Jul 25. J Cell Biol. 2005. PMID: 16043516 Free PMC article.
-
The Wlds transgene reduces axon loss in a Charcot-Marie-Tooth disease 1A rat model and nicotinamide delays post-traumatic axonal degeneration.Neurobiol Dis. 2011 Apr;42(1):1-8. doi: 10.1016/j.nbd.2010.12.006. Epub 2010 Dec 16. Neurobiol Dis. 2011. PMID: 21168501
-
Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene.Nat Neurosci. 2001 Dec;4(12):1199-206. doi: 10.1038/nn770. Nat Neurosci. 2001. PMID: 11770485
-
Molecular mechanisms in the initiation phase of Wallerian degeneration.Eur J Neurosci. 2016 Aug;44(4):2040-8. doi: 10.1111/ejn.13250. Epub 2016 May 30. Eur J Neurosci. 2016. PMID: 27062141 Review.
-
Mechanism of initiation and regulation of axonal degeneration with special reference to NMNATs and Sarm1.Neurosci Res. 2023 Dec;197:3-8. doi: 10.1016/j.neures.2021.11.002. Epub 2021 Nov 9. Neurosci Res. 2023. PMID: 34767875 Review.
Cited by
-
Elevated mitochondria-coupled NAD(P)H in endoplasmic reticulum of dopamine neurons.Mol Biol Cell. 2016 Nov 1;27(21):3214-3220. doi: 10.1091/mbc.E16-07-0479. Epub 2016 Aug 31. Mol Biol Cell. 2016. PMID: 27582392 Free PMC article.
-
The NAD(+) salvage pathway modulates cancer cell viability via p73.Cell Death Differ. 2016 Apr;23(4):669-80. doi: 10.1038/cdd.2015.134. Epub 2015 Nov 20. Cell Death Differ. 2016. PMID: 26586573 Free PMC article.
-
Intracellular redox state revealed by in vivo (31) P MRS measurement of NAD(+) and NADH contents in brains.Magn Reson Med. 2014 Jun;71(6):1959-72. doi: 10.1002/mrm.24859. Epub 2013 Jul 10. Magn Reson Med. 2014. PMID: 23843330 Free PMC article.
-
Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture.Mol Neurodegener. 2017 Oct 27;12(1):77. doi: 10.1186/s13024-017-0221-9. Mol Neurodegener. 2017. PMID: 29078798 Free PMC article.
-
Isoform-specific targeting and interaction domains in human nicotinamide mononucleotide adenylyltransferases.J Biol Chem. 2010 Jun 11;285(24):18868-76. doi: 10.1074/jbc.M110.107631. Epub 2010 Apr 13. J Biol Chem. 2010. PMID: 20388704 Free PMC article.
References
-
- Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, Luo L. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron. 2006;50:883–895. - PubMed
-
- Luo L, O'Leary DD. Axon retraction and degeneration in development and disease. Annu Rev Neurosci. 2005;28:127–156. - PubMed
-
- Waller AV. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations on the alterations produced thereby in the structure of their primitive fibres. Philosophical Transactions of the Royal Society of London. 1850;140:423–429.
-
- Schmalbruch H, Jensen HJ, Bjaerg M, Kamieniecka Z, Kurland L. A new mouse mutant with progressive motor neuronopathy. J Neuropathol Exp Neurol. 1991;50:192–204. - PubMed
-
- Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources