

A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease
- PMID: 19375058
- PMCID: PMC2681001
- DOI: 10.1016/j.ajhg.2009.03.018
A nonsense mutation in COQ9 causes autosomal-recessive neonatal-onset primary coenzyme Q10 deficiency: a potentially treatable form of mitochondrial disease
Abstract
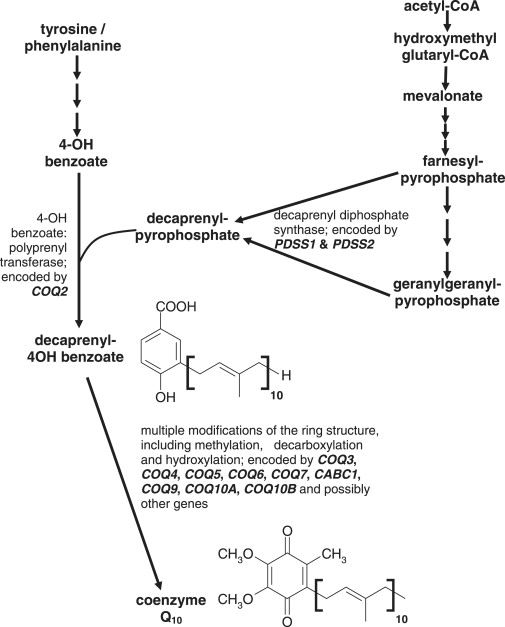
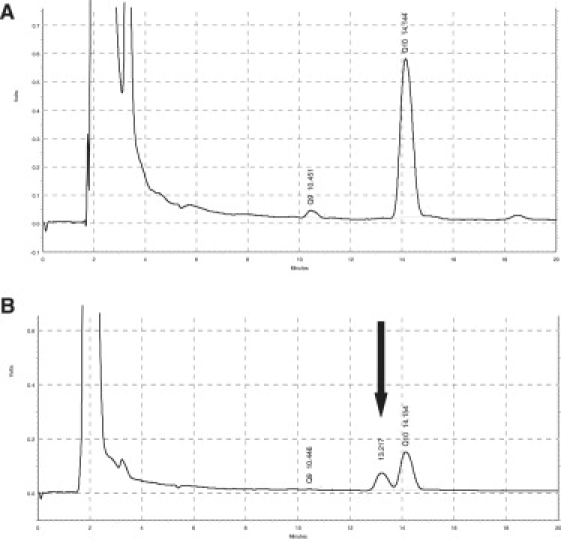
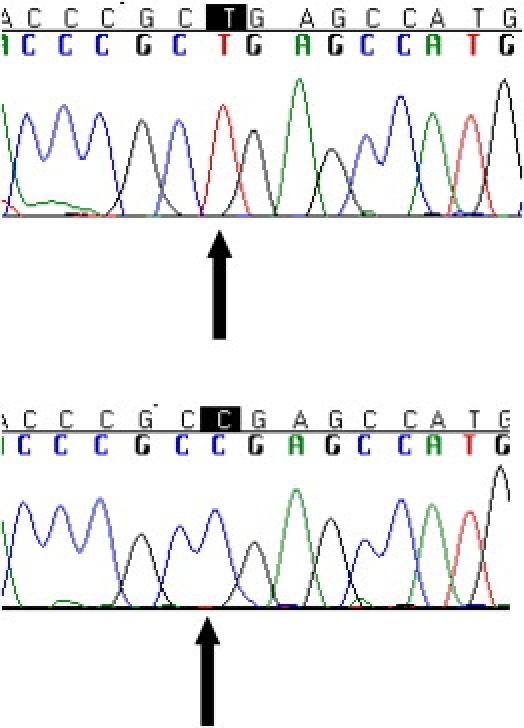
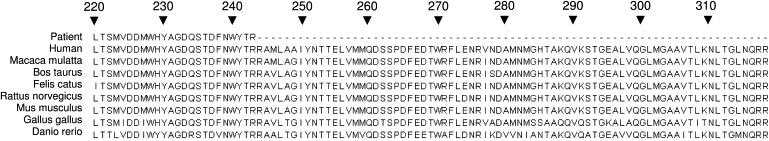
Coenzyme Q(10) is a mobile lipophilic electron carrier located in the inner mitochondrial membrane. Defects of coenzyme Q(10) biosynthesis represent one of the few treatable mitochondrial diseases. We genotyped a patient with primary coenzyme Q(10) deficiency who presented with neonatal lactic acidosis and later developed multisytem disease including intractable seizures, global developmental delay, hypertrophic cardiomyopathy, and renal tubular dysfunction. Cultured skin fibroblasts from the patient had a coenzyme Q(10) biosynthetic rate of 11% of normal controls and accumulated an abnormal metabolite that we believe to be a biosynthetic intermediate. In view of the rarity of coenzyme Q(10) deficiency, we hypothesized that the disease-causing gene might lie in a region of ancestral homozygosity by descent. Data from an Illumina HumanHap550 array were analyzed with BeadStudio software. Sixteen regions of homozygosity >1.5 Mb were identified in the affected infant. Two of these regions included the loci of two of 16 candidate genes implicated in human coenzyme Q(10) biosynthesis. Sequence analysis demonstrated a homozygous stop mutation affecting a highly conserved residue of COQ9, leading to the truncation of 75 amino acids. Site-directed mutagenesis targeting the equivalent residue in the yeast Saccharomyces cerevisiae abolished respiratory growth.
Figures
Similar articles
-
A family segregating lethal neonatal coenzyme Q10 deficiency caused by mutations in COQ9.J Inherit Metab Dis. 2018 Jul;41(4):719-729. doi: 10.1007/s10545-017-0122-7. Epub 2018 Mar 20. J Inherit Metab Dis. 2018. PMID: 29560582
-
Coenzyme Q10 deficiencies: pathways in yeast and humans.Essays Biochem. 2018 Jul 20;62(3):361-376. doi: 10.1042/EBC20170106. Print 2018 Jul 20. Essays Biochem. 2018. PMID: 29980630 Free PMC article. Review.
-
Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency.Biochim Biophys Acta. 2014 Jan;1842(1):1-6. doi: 10.1016/j.bbadis.2013.10.007. Epub 2013 Oct 18. Biochim Biophys Acta. 2014. PMID: 24140869 Free PMC article.
-
The molecular genetics of coenzyme Q biosynthesis in health and disease.Biochimie. 2014 May;100:78-87. doi: 10.1016/j.biochi.2013.12.006. Epub 2013 Dec 16. Biochimie. 2014. PMID: 24355204 Review.
-
Two cases of neonatal hyperglycemia caused by a homozygous COQ9 stop-gain variant.J Diabetes Investig. 2025 May;16(5):959-963. doi: 10.1111/jdi.70022. Epub 2025 Mar 10. J Diabetes Investig. 2025. PMID: 40062559 Free PMC article.
Cited by
-
Mitochondrial Diseases Part I: mouse models of OXPHOS deficiencies caused by defects in respiratory complex subunits or assembly factors.Mitochondrion. 2015 Mar;21:76-91. doi: 10.1016/j.mito.2015.01.009. Epub 2015 Feb 4. Mitochondrion. 2015. PMID: 25660179 Free PMC article. Review.
-
Coenzyme q10 administration in community-acquired pneumonia in the elderly.Iran Red Crescent Med J. 2014 Dec 1;16(12):e18852. doi: 10.5812/ircmj.18852. eCollection 2014 Dec. Iran Red Crescent Med J. 2014. PMID: 25763241 Free PMC article.
-
Biosynthesis, Deficiency, and Supplementation of Coenzyme Q.Antioxidants (Basel). 2023 Jul 21;12(7):1469. doi: 10.3390/antiox12071469. Antioxidants (Basel). 2023. PMID: 37508007 Free PMC article. Review.
-
Animal Models of Coenzyme Q Deficiency: Mechanistic and Translational Learnings.Antioxidants (Basel). 2021 Oct 26;10(11):1687. doi: 10.3390/antiox10111687. Antioxidants (Basel). 2021. PMID: 34829558 Free PMC article. Review.
-
Novel recessive mutations in COQ4 cause severe infantile cardiomyopathy and encephalopathy associated with CoQ10 deficiency.Mol Genet Metab Rep. 2017 May 11;12:23-27. doi: 10.1016/j.ymgmr.2017.05.001. eCollection 2017 Sep. Mol Genet Metab Rep. 2017. PMID: 28540186 Free PMC article.
References
-
- Turunen M., Olsson J., Dallner G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta. 2004;1660:171–199. - PubMed
-
- Sobreira C., Hirano M., Shanske S., Keller R.K., Haller R.G., Davidson E., Santorelli F.M., Miranda A.F., Bonilla E., Mojon D.S. Mitochondrial encephalomyopathy with coenzyme Q10 deficiency. Neurology. 1997;48:1238–1243. - PubMed
-
- Rotig A., Appelkvist E.L., Geromel V., Chretien D., Kadhom N., Edery P., Lebideau M., Dallner G., Munnich A., Ernster L. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet. 2000;356:391–395. - PubMed
-
- Rahman S., Hargreaves I., Clayton P., Heales S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001;139:456–458. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
