

Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema
- PMID: 19376887
- PMCID: PMC2711808
- DOI: 10.1152/ajplung.90406.2008
Novel critical role of Toll-like receptor 4 in lung ischemia-reperfusion injury and edema
Abstract
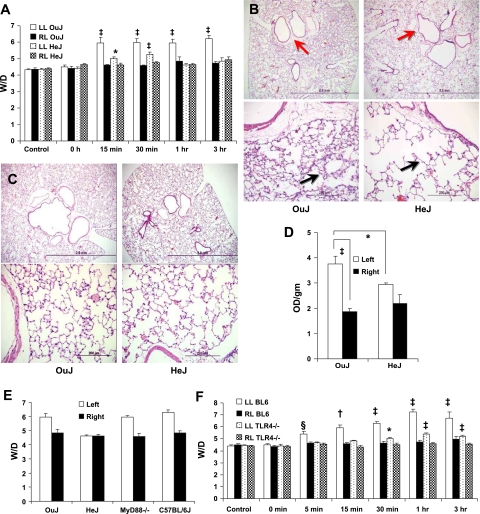
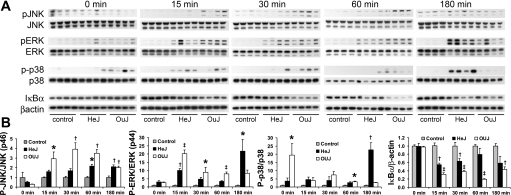
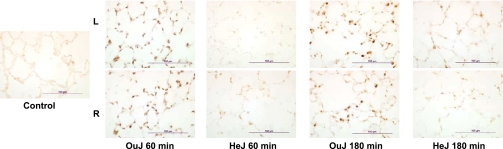
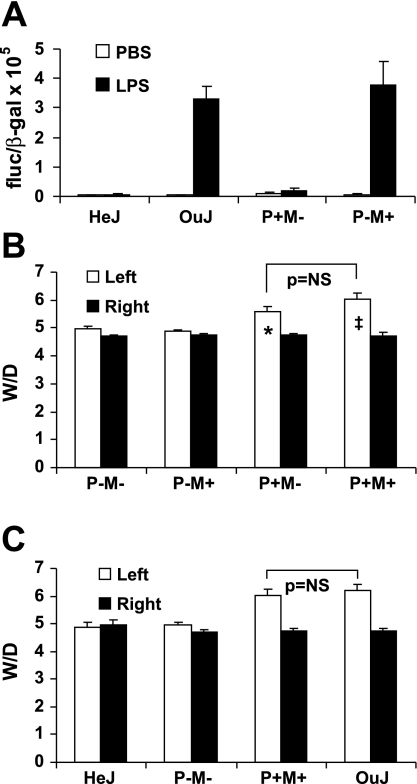
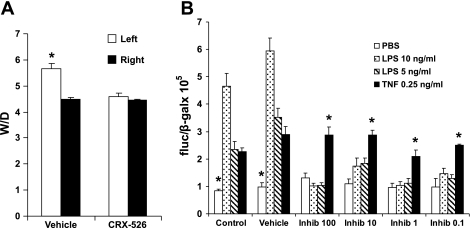
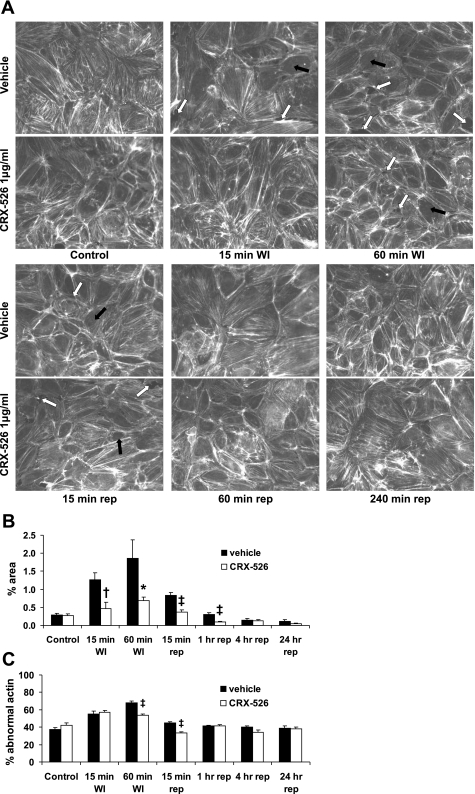
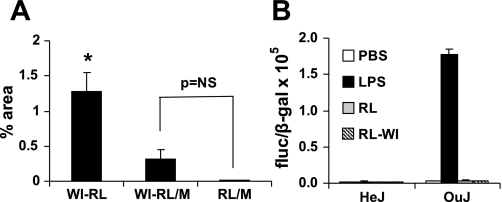
Toll-like receptors (TLRs) of the innate immune system contribute to noninfectious inflammatory processes. We employed a murine model of hilar clamping (1 h) with reperfusion times between 15 min and 3 h in TLR4-sufficient (C3H/OuJ) and TLR4-deficient (C3H/HeJ) anesthetized mice with additional studies in chimeric and myeloid differentiation factor 88 (MyD88)- and TLR4-deficient mice to determine the role of TLR4 in lung ischemia-reperfusion injury. Human pulmonary microvascular endothelial monolayers were subjected to simulated warm ischemia and reperfusion with and without CRX-526, a competitive TLR4 inhibitor. Functional TLR4 solely on pulmonary parenchymal cells, not bone marrow-derived cells, mediates early lung edema following ischemia-reperfusion independent of MyD88. Activation of MAPKs and NF-kappaB was significantly blunted and/or delayed in lungs of TLR4-deficient mice as a consequence of ischemia-reperfusion injury, but edema development appeared to be independent of activation of these signaling pathways. Pretreatment with a competitive TLR4 inhibitor prevented edema in vivo and reduced actin cytoskeletal rearrangement and gap formation in pulmonary microvascular endothelial monolayers subjected to simulated warm ischemia and reperfusion. In addition to its well-accepted role to alter gene transcription, functioning TLR4 on pulmonary parenchymal cells plays a key role in very early and profound pulmonary edema in murine lung ischemia-reperfusion injury. This may be due to a novel mechanism: regulation of endothelial cell cytoskeleton affecting microvascular endothelial cell permeability.
Figures
Similar articles
-
TLR4 mediates lung injury and inflammation in intestinal ischemia-reperfusion.J Surg Res. 2012 May 15;174(2):326-33. doi: 10.1016/j.jss.2010.12.005. Epub 2011 Jan 5. J Surg Res. 2012. PMID: 21392794
-
Ischemia-reperfusion lung injury is attenuated in MyD88-deficient mice.PLoS One. 2013 Oct 11;8(10):e77123. doi: 10.1371/journal.pone.0077123. eCollection 2013. PLoS One. 2013. PMID: 24146959 Free PMC article.
-
Dexmedetomidine protects against lung injury induced by limb ischemia-reperfusion via the TLR4/MyD88/NF-κB pathway.Kaohsiung J Med Sci. 2019 Nov;35(11):672-678. doi: 10.1002/kjm2.12115. Epub 2019 Aug 2. Kaohsiung J Med Sci. 2019. PMID: 31373750 Free PMC article.
-
Hydroxysafflor yellow A exerts neuroprotective effects in cerebral ischemia reperfusion-injured mice by suppressing the innate immune TLR4-inducing pathway.Eur J Pharmacol. 2015 Dec 15;769:324-32. doi: 10.1016/j.ejphar.2015.11.036. Epub 2015 Nov 25. Eur J Pharmacol. 2015. PMID: 26607471
-
Plasma membrane-bound G protein-coupled bile acid receptor attenuates liver ischemia/reperfusion injury via the inhibition of toll-like receptor 4 signaling in mice.Liver Transpl. 2017 Jan;23(1):63-74. doi: 10.1002/lt.24628. Liver Transpl. 2017. PMID: 27597295
Cited by
-
Advances in lung ischemia/reperfusion injury: unraveling the role of innate immunity.Inflamm Res. 2024 Mar;73(3):393-405. doi: 10.1007/s00011-023-01844-7. Epub 2024 Jan 24. Inflamm Res. 2024. PMID: 38265687 Review.
-
MicroRNA-146a-mediated downregulation of IRAK1 protects mouse and human small intestine against ischemia/reperfusion injury.EMBO Mol Med. 2012 Dec;4(12):1308-19. doi: 10.1002/emmm.201201298. Epub 2012 Nov 9. EMBO Mol Med. 2012. PMID: 23143987 Free PMC article.
-
DNA attenuates enterocyte Toll-like receptor 4-mediated intestinal mucosal injury after remote trauma.Am J Physiol Gastrointest Liver Physiol. 2011 May;300(5):G862-73. doi: 10.1152/ajpgi.00373.2010. Epub 2011 Jan 13. Am J Physiol Gastrointest Liver Physiol. 2011. PMID: 21233273 Free PMC article.
-
Animal models of ex vivo lung perfusion as a platform for transplantation research.World J Exp Med. 2014 May 20;4(2):7-15. doi: 10.5493/wjem.v4.i2.7. eCollection 2014 May 20. World J Exp Med. 2014. PMID: 24977117 Free PMC article. Review.
-
Mechanical ventilation modulates Toll-like receptors 2, 4, and 9 on alveolar macrophages in a ventilator-induced lung injury model.J Thorac Dis. 2015 Apr;7(4):616-24. doi: 10.3978/j.issn.2072-1439.2015.02.10. J Thorac Dis. 2015. PMID: 25973227 Free PMC article.
References
-
- Casiraghi M, Abano J, Burridge K, Randell S, Egan T. Toll-like receptor-4 (TLR4) inhibition reduces actin cytoskeletal re-arrangement and gap formation in cultured human pulmonary microvascular endothelial cells (HMVECs) subjected to simulated warm ischemia-reperfusion injury (IRI) (Abstract). J Heart Lung Transplant 27: S214–S215, 2008.
-
- Cho HY, Kleeberger SR. Genetic mechanisms of susceptibility to oxidative lung injury in mice. Free Radic Biol Med 42: 433–445, 2007. - PubMed
-
- Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem 274: 10689–10692, 1999. - PubMed
-
- de Groot H, Rauen U. Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplant Proc 39: 481–484, 2007. - PubMed
-
- de Perrot M, Liu M, Waddell TK, Keshavjee S. Ischemia-reperfusion-induced lung injury. Am J Respir Crit Care Med 167: 490–511, 2003. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous