

Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize
- PMID: 19376930
- PMCID: PMC2685623
- DOI: 10.1105/tpc.109.065714
Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize
Abstract
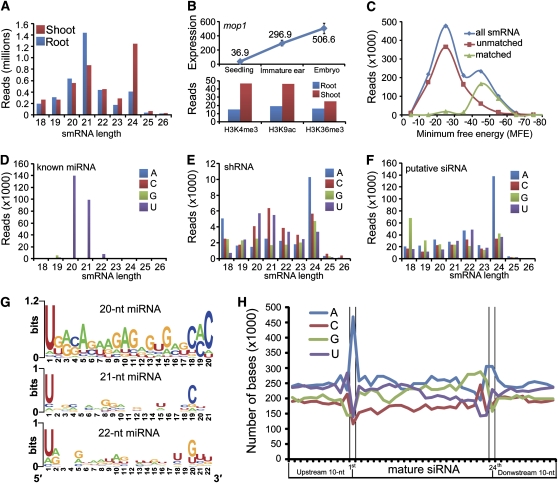
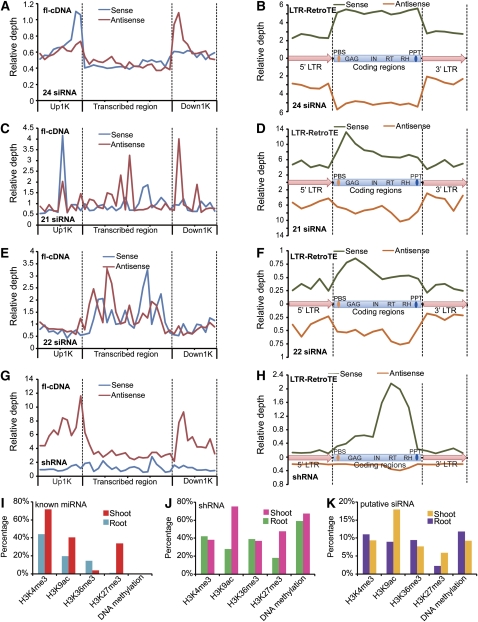
Maize (Zea mays) has an exceptionally complex genome with a rich history in both epigenetics and evolution. We report genomic landscapes of representative epigenetic modifications and their relationships to mRNA and small RNA (smRNA) transcriptomes in maize shoots and roots. The epigenetic patterns differed dramatically between genes and transposable elements, and two repressive marks (H3K27me3 and DNA methylation) were usually mutually exclusive. We found an organ-specific distribution of canonical microRNAs (miRNAs) and endogenous small interfering RNAs (siRNAs), indicative of their tissue-specific biogenesis. Furthermore, we observed that a decreasing level of mop1 led to a concomitant decrease of 24-nucleotide siRNAs relative to 21-nucleotide miRNAs in a tissue-specific manner. A group of 22-nucleotide siRNAs may originate from long-hairpin double-stranded RNAs and preferentially target gene-coding regions. Additionally, a class of miRNA-like smRNAs, whose putative precursors can form short hairpins, potentially targets genes in trans. In summary, our data provide a critical analysis of the maize epigenome and its relationships to mRNA and smRNA transcriptomes.
Figures
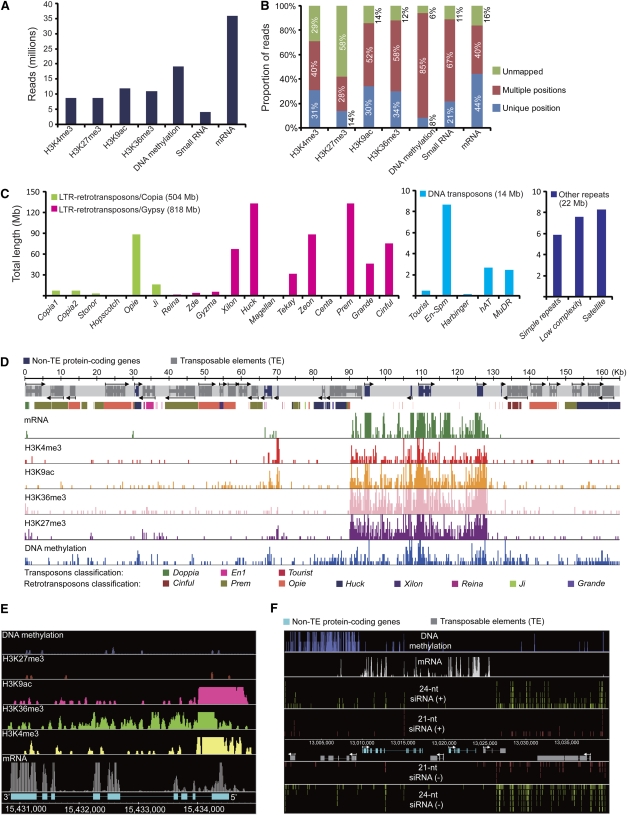
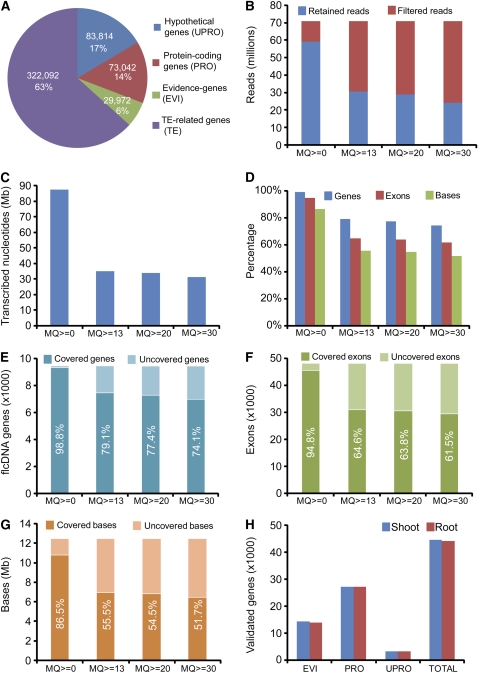
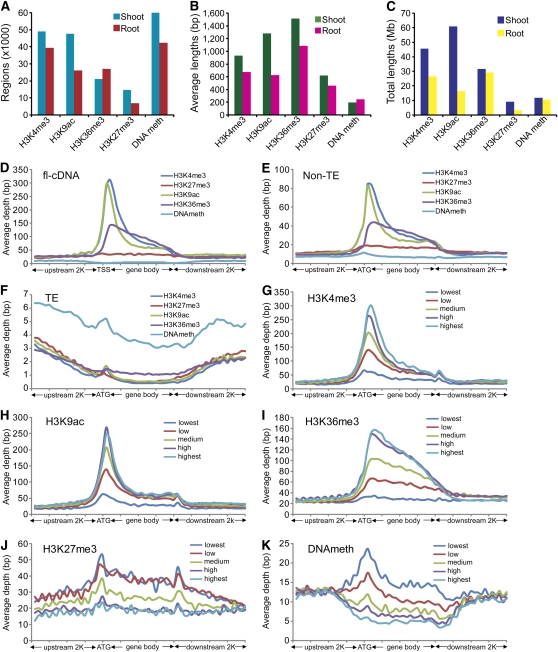
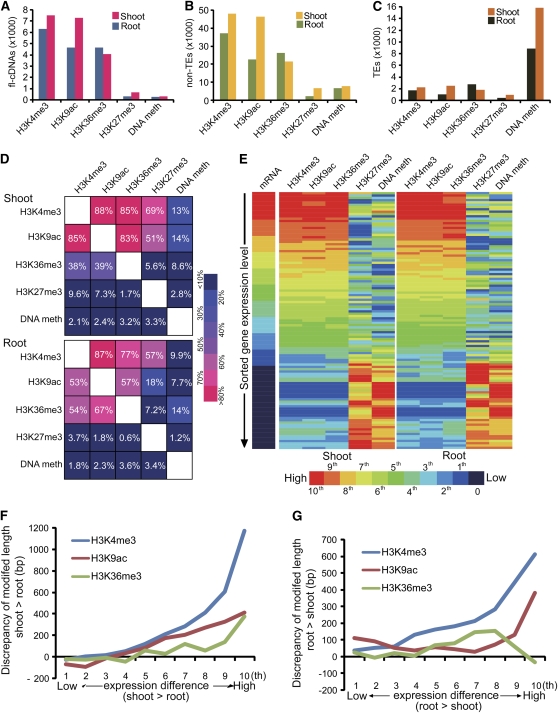

Comment in
-
Deep sequencing maps the maize epigenomic landscape.Plant Cell. 2009 Apr;21(4):1024-6. doi: 10.1105/tpc.109.068064. Plant Cell. 2009. PMID: 19557884 Free PMC article. No abstract available.
-
Next-generation sequencing reveals complex relationships between the epigenome and transcriptome in maize.Plant Signal Behav. 2009 Aug;4(8):760-2. doi: 10.1105/tpc.109.065714. Epub 2009 Aug 3. Plant Signal Behav. 2009. PMID: 19820310 Free PMC article.
References
-
- Barski, A., Cuddapah, S., Cui, K., Roh, T.Y., Schones, D.E., Wang, Z., Wei, G., Chepelev, I., and Zhao, K. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129 823–837. - PubMed
-
- Bernstein, B.E., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 315–326. - PubMed
-
- Bonnet, E., Van de Peer, Y., and Rouzé, P. (2006). The small RNA world of plants. New Phytol. 171 451–468. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
