

PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB
- PMID: 19376972
- PMCID: PMC2678648
- DOI: 10.1073/pnas.0806742106
PPARalpha blocks glucocorticoid receptor alpha-mediated transactivation but cooperates with the activated glucocorticoid receptor alpha for transrepression on NF-kappaB
Abstract
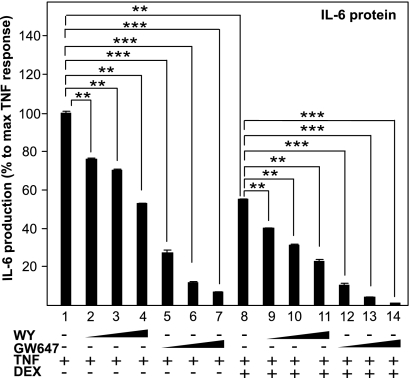
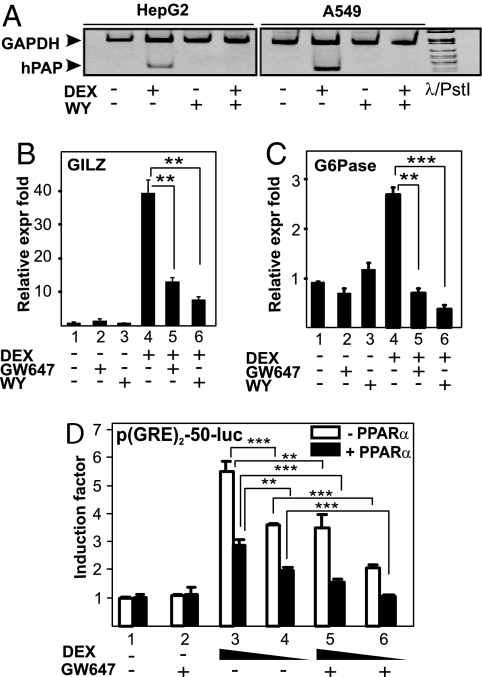
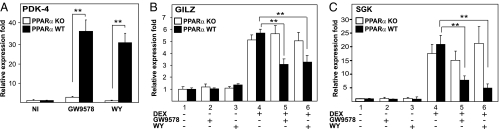
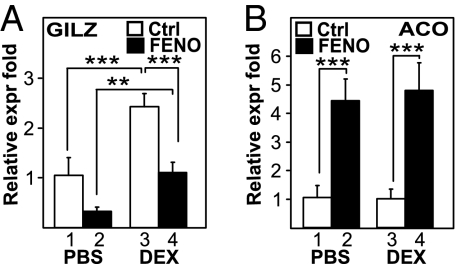
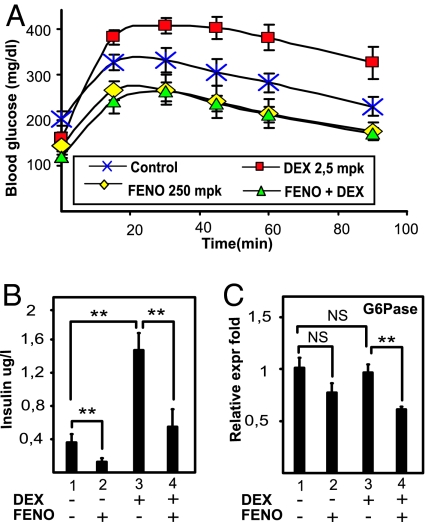
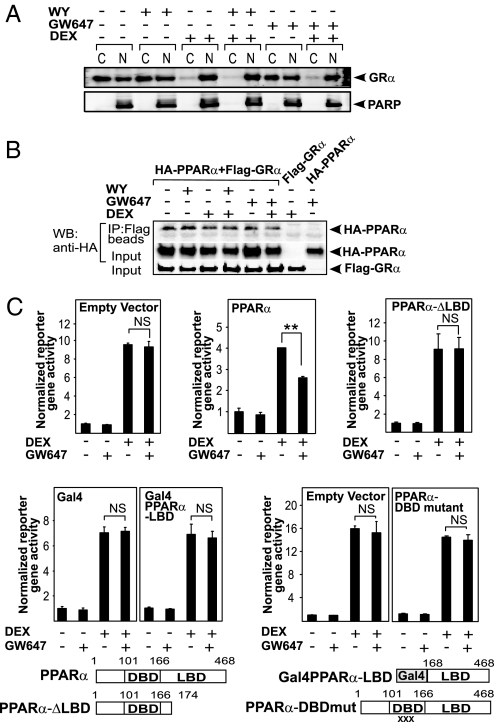
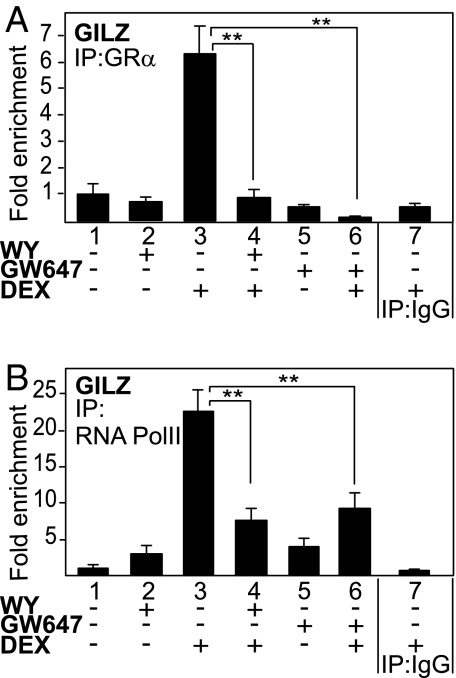
Glucocorticoid receptor alpha (GRalpha) and peroxisome proliferator-activated receptor alpha (PPARalpha) are transcription factors with clinically important immune-modulating properties. Either receptor can inhibit cytokine gene expression, mainly through interference with nuclear factor kappaB (NF-kappaB)-driven gene expression. The present work aimed to investigate a functional cross-talk between PPARalpha- and GRalpha-mediated signaling pathways. Simultaneous activation of PPARalpha and GRalpha dose-dependently enhances transrepression of NF-kappaB-driven gene expression and additively represses cytokine production. In sharp contrast and quite unexpectedly, PPARalpha agonists inhibit the expression of classical glucocorticoid response element (GRE)-driven genes in a PPARalpha-dependent manner, as demonstrated by experiments using PPARalpha wild-type and knockout mice. The underlying mechanism for this transcriptional antagonism relies on a PPARalpha-mediated interference with the recruitment of GRalpha, and concomitantly of RNA polymerase II, to GRE-driven gene promoters. Finally, the biological relevance of this phenomenon is underscored by the observation that treatment with the PPARalpha agonist fenofibrate prevents glucocorticoid-induced hyperinsulinemia of mice fed a high-fat diet. Taken together, PPARalpha negatively interferes with GRE-mediated GRalpha activity while potentiating its antiinflammatory effects, thus providing a rationale for combination therapy in chronic inflammatory disorders.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the GR and NF-κB or AP-1: Molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. - PubMed
-
- Shiri-Sverdlov R, et al. Early diet-induced non-alcoholic steatohepatitis in APOE2 knockin mice and its prevention by fibrates. J Hepatol. 2006;44:732–741. - PubMed
-
- Desvergne B, Wahli W. PPARs: Nuclear control of metabolism. Endocr Rev. 1999;20:649–688. - PubMed
-
- Delerive P, et al. PPARα negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–32054. - PubMed
-
- Staels B, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998;393:790–793. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
