

T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model
- PMID: 19377047
- PMCID: PMC2710932
- DOI: 10.1182/blood-2009-03-209650
T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model
Abstract
For the adoptive transfer of tumor-directed T lymphocytes to prove effective, there will probably need to be a match between the chemokines the tumor produces and the chemokine receptors the effector T cells express. The Reed-Stemberg cells of Hodgkin lymphoma (HL) predominantly produce thymus- and activation-regulated chemokine/CC chemokine ligand 17 (TARC/CCL17) and macrophage-derived chemokine (MDC/CCL22), which preferentially attract type 2 T helper (Th2) cells and regulatory T cells (Tregs) that express the TARC/MDC-specific chemokine receptor CCR4, thus generating an immunosuppressed tumor environment. By contrast, effector CD8(+) T cells lack CCR4, are nonresponsive to these chemokines and are rarely detected at the tumor site. We now show that forced expression of CCR4 by effector T cells enhances their migration to HL cells. Furthermore, T lymphocytes expressing both CCR4 and a chimeric antigen receptor directed to the HL associated antigen CD30 sustain their cytotoxic function and cytokine secretion in vitro, and produce enhanced tumor control when infused intravenously in mice engrafted with human HL. This approach may be of value in patients affected by HL.
Figures
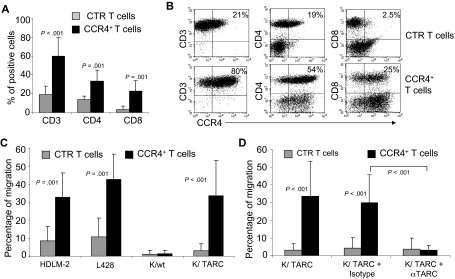
 represent the mean ± SD of control T cells; ■, mean ± SD of CCR4+ T cells. The data summarize the results of T-cell lines generated from 7 healthy donors. (B) A representative phenotypic analysis. (C) The migration of CTR () and CCR4+ (■) T cells toward TARC gradients, using a transwell migration assay. T-cell migration was evaluated using culture supernatants collected from 2 HL-derived cell lines (HDLM-2 and L428) that physiologically produce high amounts of TARC, and against the Karpas-299 cell line genetically modified to produce TARC (K/TARC). K/wt was used as a control. The panel indicates that migration toward TARC is significantly improved if T cells are genetically modified to overexpress CCR4. The data are the mean ± SD for T-cell lines generated from 7 healthy donors. (D) The improved migration of CCR4+ T cells (■) is TARC mediated as it is inhibited by addition of anti-TARC antibodies but not by the addition of an isotype control.
represent the mean ± SD of control T cells; ■, mean ± SD of CCR4+ T cells. The data summarize the results of T-cell lines generated from 7 healthy donors. (B) A representative phenotypic analysis. (C) The migration of CTR () and CCR4+ (■) T cells toward TARC gradients, using a transwell migration assay. T-cell migration was evaluated using culture supernatants collected from 2 HL-derived cell lines (HDLM-2 and L428) that physiologically produce high amounts of TARC, and against the Karpas-299 cell line genetically modified to produce TARC (K/TARC). K/wt was used as a control. The panel indicates that migration toward TARC is significantly improved if T cells are genetically modified to overexpress CCR4. The data are the mean ± SD for T-cell lines generated from 7 healthy donors. (D) The improved migration of CCR4+ T cells (■) is TARC mediated as it is inhibited by addition of anti-TARC antibodies but not by the addition of an isotype control.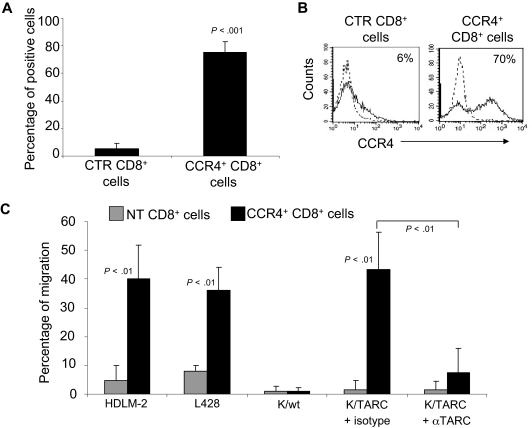 ) and CCR4+ (■) CD8+ T cells toward TARC gradients, using a transwell migration assay. The panel indicates that migration toward TARC is significantly increased when CD8+ T cells overexpress CCR4. The data are the mean ± SD for CD8+ T-cell lines generated from 4 healthy donors.
) and CCR4+ (■) CD8+ T cells toward TARC gradients, using a transwell migration assay. The panel indicates that migration toward TARC is significantly increased when CD8+ T cells overexpress CCR4. The data are the mean ± SD for CD8+ T-cell lines generated from 4 healthy donors.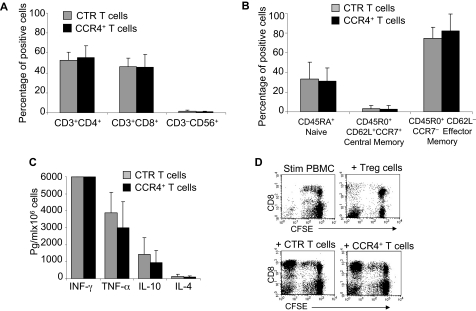 ) and CCR4+ (■) T cells. The data are mean ± SD of 5 healthy donors. T-cell markers are shown on the x-axis. No significant differences were observed if cells overexpressed CCR4. (B) Expression of naive, central memory, and effector memory surface markers on CTR () and CCR4+ (■) T cells is not significantly different. The data are mean ± SD of 4 donors. (C) The production of Th1 (IFN-γ and TNF-α) and Th2 (IL-10 and IL-4) cytokines by CTR () and CCR4+ (■) T cells 24 hours after stimulation with OKT3. No significant differences in cytokine production were detected, suggesting that the transgenic expression of CCR4 does not induce the acquisition of a Th2 profile. (D) CCR4+ T cells do not acquire inhibitory function. The inhibitory activity of T cells was evaluated using a CFSE-based suppression assay in which PBMCs labeled with CFSE are stimulated with irradiated allogeneic PBMCs and OKT3 in the absence (top left graph) or in the presence of freshly isolated CD4+CD25bright cells (top right graph), CTR (bottom left graph), or CCR4+ T cells (bottom right graph). The panel indicates a significant number of divisions (CFSE partitioning) of T cells in the absence or presence of CTR or CCR4+ T cells (evident in the left quadrants). In contrast, the divisions are significantly reduced in the presence of Treg cells. Shown is one of 3 donors studied, illustrative of results from all.
) and CCR4+ (■) T cells. The data are mean ± SD of 5 healthy donors. T-cell markers are shown on the x-axis. No significant differences were observed if cells overexpressed CCR4. (B) Expression of naive, central memory, and effector memory surface markers on CTR () and CCR4+ (■) T cells is not significantly different. The data are mean ± SD of 4 donors. (C) The production of Th1 (IFN-γ and TNF-α) and Th2 (IL-10 and IL-4) cytokines by CTR () and CCR4+ (■) T cells 24 hours after stimulation with OKT3. No significant differences in cytokine production were detected, suggesting that the transgenic expression of CCR4 does not induce the acquisition of a Th2 profile. (D) CCR4+ T cells do not acquire inhibitory function. The inhibitory activity of T cells was evaluated using a CFSE-based suppression assay in which PBMCs labeled with CFSE are stimulated with irradiated allogeneic PBMCs and OKT3 in the absence (top left graph) or in the presence of freshly isolated CD4+CD25bright cells (top right graph), CTR (bottom left graph), or CCR4+ T cells (bottom right graph). The panel indicates a significant number of divisions (CFSE partitioning) of T cells in the absence or presence of CTR or CCR4+ T cells (evident in the left quadrants). In contrast, the divisions are significantly reduced in the presence of Treg cells. Shown is one of 3 donors studied, illustrative of results from all.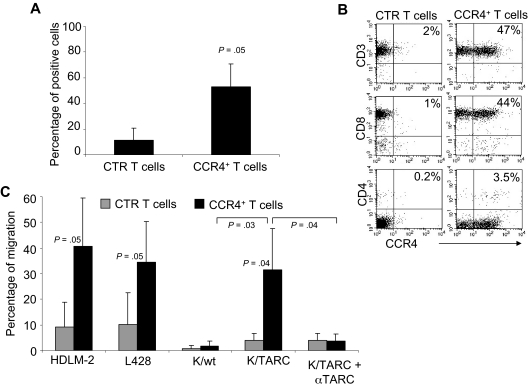 ) and transduced (■) T cells established from 4 persons with HL. The data are mean ± SD. (B) A representative phenotypic analysis. (C) The migration of CTR () and CCR4+ (■) T cells toward TARC gradients, using a transwell migration assay. T-cell migration was evaluated using culture supernatants collected from HDLM-2 and L428, which physiologically produce high amounts of TARC, and against Karpas genetically modified to produce TARC (K/TARC). K/wt represents migration toward Karpas wild-type, which produces a negligible amount of TARC, as a negative control. Migration is significantly increased for CCR4+ T cells compared with CTR T cells. In addition, the figure shows that the improved migration of CCR4+ T cells (■) is TARC mediated as it is inhibited by addition of anti-TARC antibodies but not by addition of an isotype control.
) and transduced (■) T cells established from 4 persons with HL. The data are mean ± SD. (B) A representative phenotypic analysis. (C) The migration of CTR () and CCR4+ (■) T cells toward TARC gradients, using a transwell migration assay. T-cell migration was evaluated using culture supernatants collected from HDLM-2 and L428, which physiologically produce high amounts of TARC, and against Karpas genetically modified to produce TARC (K/TARC). K/wt represents migration toward Karpas wild-type, which produces a negligible amount of TARC, as a negative control. Migration is significantly increased for CCR4+ T cells compared with CTR T cells. In addition, the figure shows that the improved migration of CCR4+ T cells (■) is TARC mediated as it is inhibited by addition of anti-TARC antibodies but not by addition of an isotype control.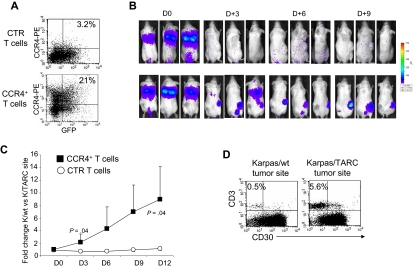
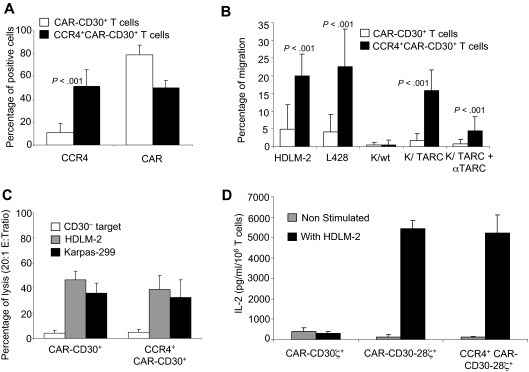 ; Karpas, ■) and CD30− (□) tumor cells by CAR-CD30+ and CCR4+CD30-CAR+ T cells. (D) The measurement of IL-2 cytokine released in the supernatant of T cells cocultured with or without HDLM-2 and assessed using a specific ELISA assay. T cells were transduced with either CAR-CD30 or CCR4(I)CAR-CD30, where CAR molecules also incorporate the CD28 endodomain. As a control, T cells were also transduced with the same CAR targeting CD30 but lacking the CD28 endodomain (CD30CARζ). As anticipated, we observed enhanced production of IL-2 by T cells transduced with the CAR containing the CD28 endodomain (CAR-CD30), regardless of coexpression of CCR4, but not by T cells transduced with CD30CARζ lacking CD28. The figure indicates that T cells transduced with CCR4(I)CAR-CD30 vector produce IL-2 in amounts comparable with that of T cells transduced with the vector encoding CAR-CD30 alone, confirming that the CD28 pathway is not impaired by the coexpression of CCR4. Data are mean ± SD of 10 donors.
; Karpas, ■) and CD30− (□) tumor cells by CAR-CD30+ and CCR4+CD30-CAR+ T cells. (D) The measurement of IL-2 cytokine released in the supernatant of T cells cocultured with or without HDLM-2 and assessed using a specific ELISA assay. T cells were transduced with either CAR-CD30 or CCR4(I)CAR-CD30, where CAR molecules also incorporate the CD28 endodomain. As a control, T cells were also transduced with the same CAR targeting CD30 but lacking the CD28 endodomain (CD30CARζ). As anticipated, we observed enhanced production of IL-2 by T cells transduced with the CAR containing the CD28 endodomain (CAR-CD30), regardless of coexpression of CCR4, but not by T cells transduced with CD30CARζ lacking CD28. The figure indicates that T cells transduced with CCR4(I)CAR-CD30 vector produce IL-2 in amounts comparable with that of T cells transduced with the vector encoding CAR-CD30 alone, confirming that the CD28 pathway is not impaired by the coexpression of CCR4. Data are mean ± SD of 10 donors.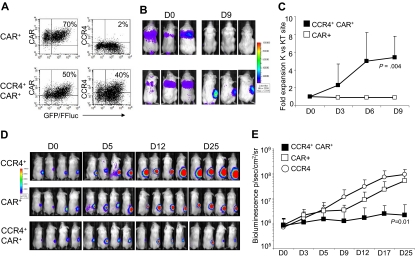
Comment in
-
Chemokine receptors as therapeutic tools in Hodgkin lymphoma: CCR4 and beyond.Blood. 2010 Jan 21;115(3):746-7; author reply 748. doi: 10.1182/blood-2009-10-247809. Blood. 2010. PMID: 20093413 No abstract available.
References
-
- Roskrow MA, Suzuki N, Gan Y, et al. Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of patients with EBV-positive relapsed Hodgkin's disease. Blood. 1998;91:2925–2934. - PubMed
-
- Wu TC, Mann RB, Charache P, et al. Detection of EBV gene expression in Reed-Sternberg cells of Hodgkin's disease. Int J Cancer. 1990;46:801–804. - PubMed
-
- Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85:1–14. - PubMed
-
- Hombach A, Heuser C, Sircar R, et al. An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin's lymphoma cells in the presence of soluble CD30. Cancer Res. 1998;58:1116–1119. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
