

FastTree: computing large minimum evolution trees with profiles instead of a distance matrix
- PMID: 19377059
- PMCID: PMC2693737
- DOI: 10.1093/molbev/msp077
FastTree: computing large minimum evolution trees with profiles instead of a distance matrix
Abstract
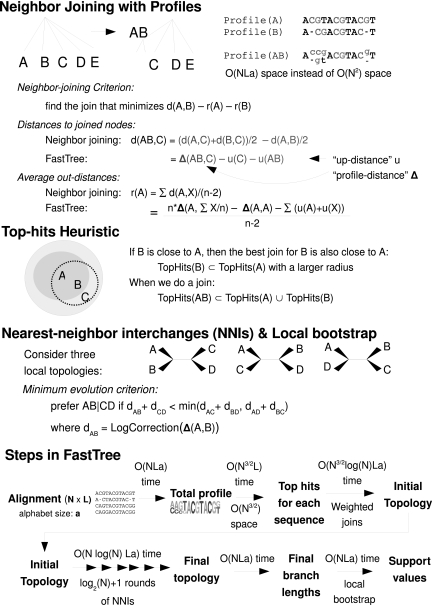
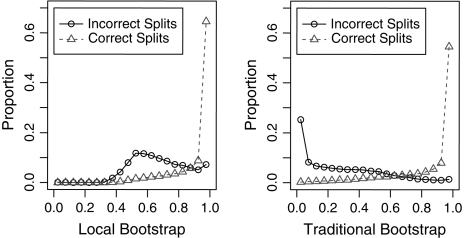
Gene families are growing rapidly, but standard methods for inferring phylogenies do not scale to alignments with over 10,000 sequences. We present FastTree, a method for constructing large phylogenies and for estimating their reliability. Instead of storing a distance matrix, FastTree stores sequence profiles of internal nodes in the tree. FastTree uses these profiles to implement Neighbor-Joining and uses heuristics to quickly identify candidate joins. FastTree then uses nearest neighbor interchanges to reduce the length of the tree. For an alignment with N sequences, L sites, and a different characters, a distance matrix requires O(N(2)) space and O(N(2)L) time, but FastTree requires just O(NLa + N ) memory and O(N log (N)La) time. To estimate the tree's reliability, FastTree uses local bootstrapping, which gives another 100-fold speedup over a distance matrix. For example, FastTree computed a tree and support values for 158,022 distinct 16S ribosomal RNAs in 17 h and 2.4 GB of memory. Just computing pairwise Jukes-Cantor distances and storing them, without inferring a tree or bootstrapping, would require 17 h and 50 GB of memory. In simulations, FastTree was slightly more accurate than Neighbor-Joining, BIONJ, or FastME; on genuine alignments, FastTree's topologies had higher likelihoods. FastTree is available at http://microbesonline.org/fasttree.
Figures
References
-
- Bininda-Emonds OR, Brady SG, Kim J, Sanderson MJ. Scaling of accuracy in extremely large phylogenetic trees. Pac Symp Biocomput. 2001;2001:547–558. - PubMed
-
- DeLong ER, Clarke-Pearson DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1998;44:837–845. - PubMed
-
- Desper R, Gascuel O. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J Comput Biol. 2002;9:687–705. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
