

Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues
- PMID: 19382883
- PMCID: PMC5117106
- DOI: 10.1517/17425240902824808
Prodrug approaches to improving the oral absorption of antiviral nucleotide analogues
Abstract
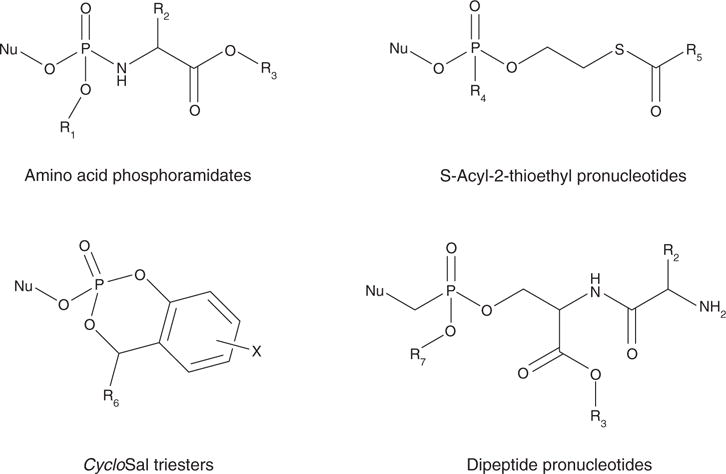
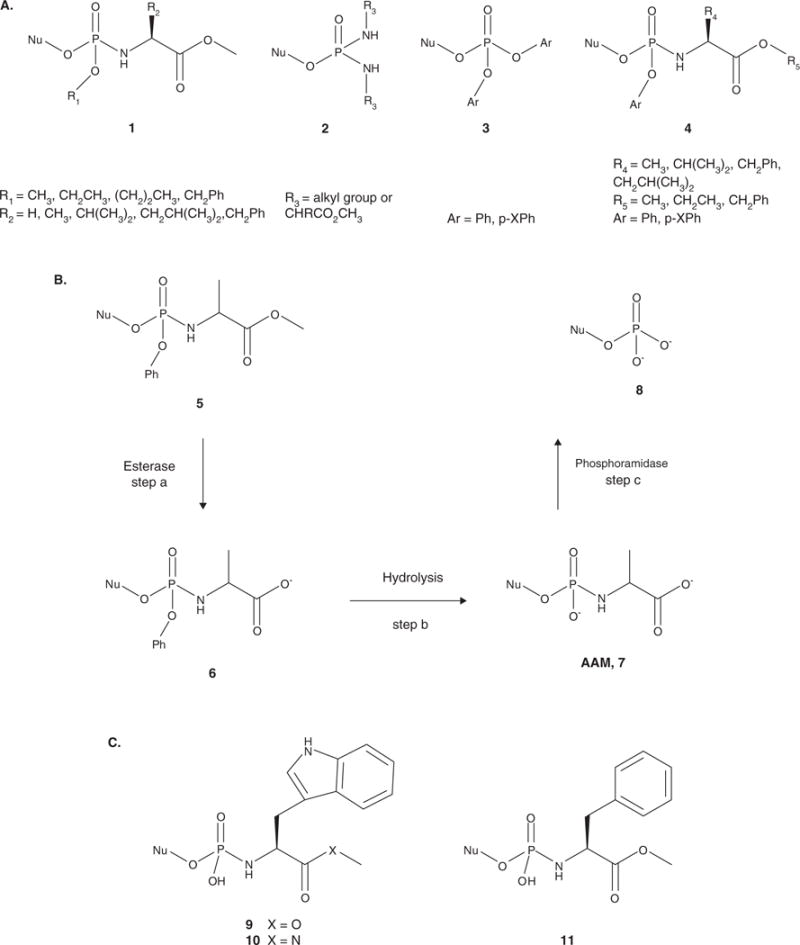
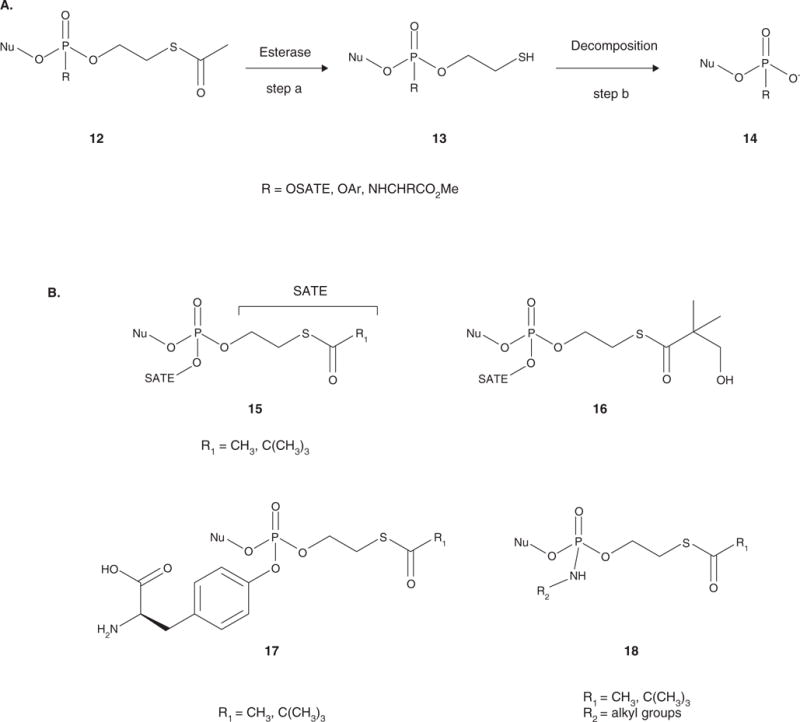
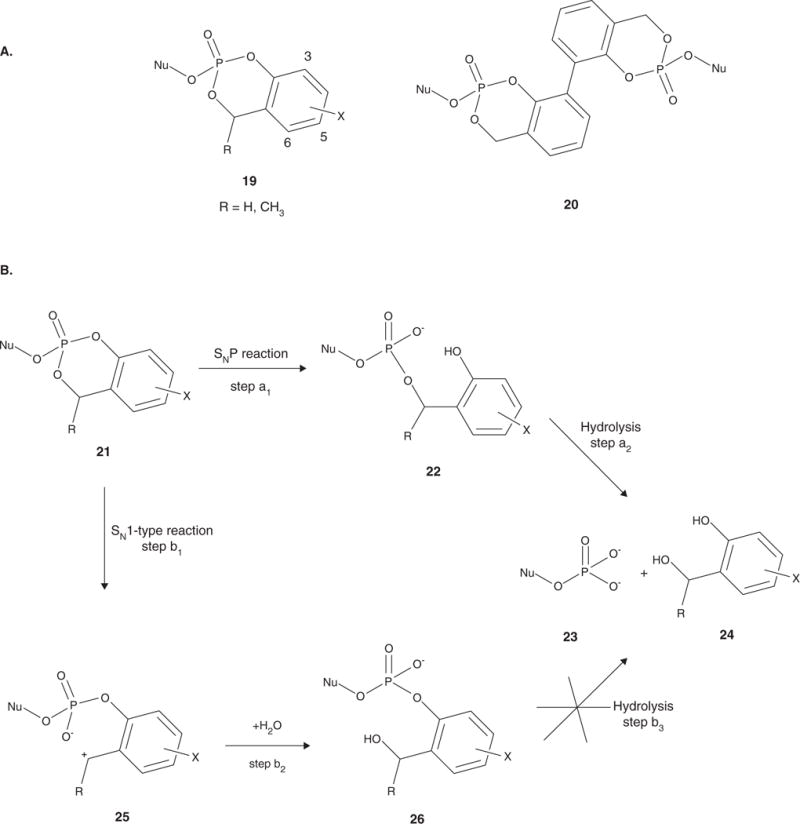
Nucleotide analogues have been well accepted as therapeutic agents active against a number of viruses. However, their use as antiviral agents is limited by the need for phosphorylation by endogenous enzymes, and if the analogue is orally administered, by low bioavailability due to the presence of an ionizable diacid group. To circumvent these limitations, a number of prodrug approaches have been proposed. The ideal prodrug achieves delivery of a parent drug by attachment of a non-toxic moiety that is stable during transport and delivery, but is readily cleaved to release the parent drug once at the target. Here, a brief overview of several promising prodrug strategies currently under development is given.
Conflict of interest statement
Declaration of interest LP and CM are co-inventors on a patent related to a portion of the work discussed in this review.
Figures
Similar articles
-
Nucleotide Prodrug Containing a Nonproteinogenic Amino Acid To Improve Oral Delivery of a Hepatitis C Virus Treatment.Antimicrob Agents Chemother. 2018 Jul 27;62(8):e00620-18. doi: 10.1128/AAC.00620-18. Print 2018 Aug. Antimicrob Agents Chemother. 2018. PMID: 29866875 Free PMC article.
-
HepDirect prodrugs for targeting nucleotide-based antiviral drugs to the liver.Curr Opin Investig Drugs. 2006 Feb;7(2):109-17. Curr Opin Investig Drugs. 2006. PMID: 16499280 Review.
-
Prodrug strategies for improved efficacy of nucleoside antiviral inhibitors.Curr Opin HIV AIDS. 2013 Nov;8(6):556-64. doi: 10.1097/COH.0000000000000007. Curr Opin HIV AIDS. 2013. PMID: 24100876 Free PMC article. Review.
-
Enhanced oral bioavailability of DDI after administration of 6-Cl-ddP, an adenosine deaminase-activated prodrug, to chronically catheterized rats.Pharm Res. 1995 Aug;12(8):1126-33. doi: 10.1023/a:1016299507382. Pharm Res. 1995. PMID: 7494823
-
To be drug or prodrug: structure-property exploratory approach regarding oral bioavailability.J Pharm Pharm Sci. 2014;17(4):532-40. doi: 10.18433/j3bs4h. J Pharm Pharm Sci. 2014. PMID: 25579432
Cited by
-
Chemoenzymatic syntheses and anti-HIV-1 activity of glucose-nucleoside conjugates as prodrugs.Bioconjug Chem. 2010 Dec 15;21(12):2239-49. doi: 10.1021/bc1002168. Epub 2010 Nov 15. Bioconjug Chem. 2010. PMID: 21077659 Free PMC article.
-
Synthesis of 4-thiouridines with prodrug functionalization for RNA metabolic labeling.RSC Chem Biol. 2022 Feb 25;3(4):447-455. doi: 10.1039/d2cb00001f. eCollection 2022 Apr 6. RSC Chem Biol. 2022. PMID: 35441143 Free PMC article.
-
Photoactivatable prodrug for simultaneous release of mertansine and CO along with a BODIPY derivative as a luminescent marker in mitochondria: a proof of concept for NIR image-guided cancer therapy.Chem Sci. 2020 Dec 23;12(7):2667-2673. doi: 10.1039/d0sc06270g. Chem Sci. 2020. PMID: 34164035 Free PMC article.
-
Tyrosine-based 1-(S)-[3-hydroxy-2-(phosphonomethoxy)propyl]cytosine and -adenine ((S)-HPMPC and (S)-HPMPA) prodrugs: synthesis, stability, antiviral activity, and in vivo transport studies.J Med Chem. 2011 Aug 25;54(16):5680-93. doi: 10.1021/jm2001426. Epub 2011 Aug 3. J Med Chem. 2011. PMID: 21812420 Free PMC article.
-
Fludarabine Inhibits Infection of Zika Virus, SFTS Phlebovirus, and Enterovirus A71.Viruses. 2021 Apr 27;13(5):774. doi: 10.3390/v13050774. Viruses. 2021. PMID: 33925713 Free PMC article.
References
-
- De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30(2):115–33. - PubMed
-
- Rideout JL, Henry DW, Beacham LM III, editors. Nucleosides, nucleotides, and their biological applications. (Proceedings of the 5th International Round Table, October 20–22, 1982) Academic Press; New York: 1983.
-
- Ariza ME. Current prodrug strategies for the delivery of nucleotides into cells. Drug Des Rev Online. 2005;2(5):373–87. A thorough review of current prodrug strategies as utilized with nucleotides for all purposes.
-
- Hostetler KY. Synthesis and antiviral evaluation of broad spectrum, orally active analogs of cidofovir and other acyclic nucleoside phosphonates. Adv Antivir Drug Des. 2007;5:167–84.
-
- Li F, Maag H, Alfredson T. Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J Pharm Sci. 2008;97(3):1109–34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources