

Transmembrane helical domain of the cannabinoid CB1 receptor
- PMID: 19383469
- PMCID: PMC2718272
- DOI: 10.1016/j.bpj.2008.12.3934
Transmembrane helical domain of the cannabinoid CB1 receptor
Abstract
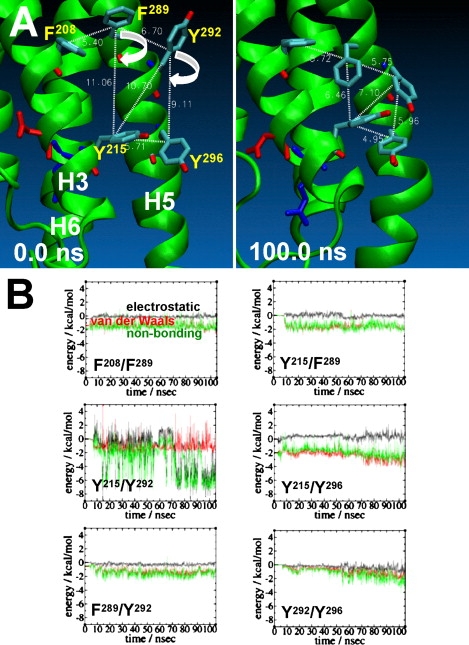
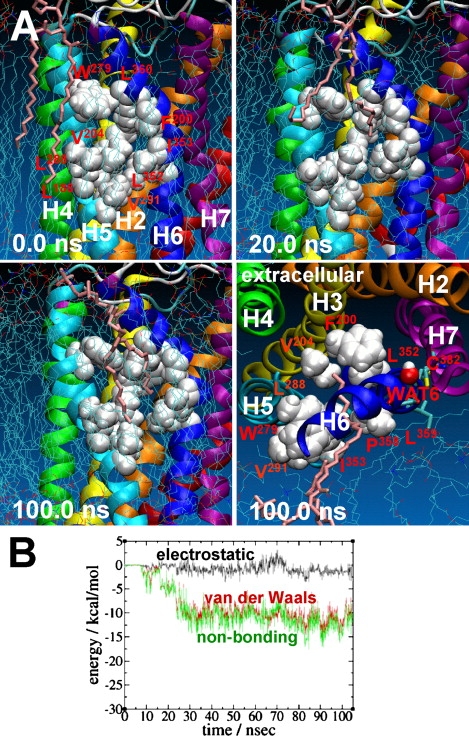
Brain cannabinoid (CB(1)) receptors are G-protein coupled receptors and belong to the rhodopsin-like subfamily. A homology model of the inactive state of the CB(1) receptor was constructed using the x-ray structure of beta(2)-adrenergic receptor (beta(2)AR) as the template. We used 105 ns duration molecular-dynamics simulations of the CB(1) receptor embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer to gain some insight into the structure and function of the CB(1) receptor. As judged from the root mean-square deviations combined with the detailed structural analyses, the helical bundle of the CB(1) receptor appears to be fully converged in 50 ns of the simulation. The results reveal that the helical bundle structure of the CB(1) receptor maintains a topology quite similar to the x-ray structures of G-protein coupled receptors overall. It is also revealed that the CB(1) receptor is stabilized by the formation of extensive, water-mediated H-bond networks, aromatic stacking interactions, and receptor-lipid interactions within the helical core region. It is likely that these interactions, which are often specific to functional motifs, including the S(N)LAxAD, D(E)RY, CWxP, and NPxxY motifs, are the molecular constraints imposed on the inactive state of the CB(1) receptor. It appears that disruption of these specific interactions is necessary to release the molecular constraints to achieve a conformational change of the receptor suitable for G-protein activation.
Figures




Similar articles
-
Understanding functional residues of the cannabinoid CB1.Curr Top Med Chem. 2010;10(8):779-98. doi: 10.2174/156802610791164210. Curr Top Med Chem. 2010. PMID: 20370713 Free PMC article. Review.
-
The cannabinoid type-1 receptor carboxyl-terminus, more than just a tail.J Neurochem. 2011 Apr;117(1):1-18. doi: 10.1111/j.1471-4159.2011.07186.x. Epub 2011 Feb 11. J Neurochem. 2011. PMID: 21244428 Free PMC article. Review.
-
Insights into the conformational perturbations of novel agonists with β3-adrenergic receptor using molecular dynamics simulations.Biochimie. 2014 Jun;101:168-82. doi: 10.1016/j.biochi.2014.01.016. Epub 2014 Feb 6. Biochimie. 2014. PMID: 24508605
-
Modeling of ligand binding to G protein coupled receptors: cannabinoid CB1, CB2 and adrenergic β 2 AR.J Mol Model. 2011 Sep;17(9):2353-66. doi: 10.1007/s00894-011-0986-7. Epub 2011 Mar 2. J Mol Model. 2011. PMID: 21365223
-
Distinct second extracellular loop structures of the brain cannabinoid CB(1) receptor: implication in ligand binding and receptor function.Proteins. 2011 Feb;79(2):581-97. doi: 10.1002/prot.22907. Proteins. 2011. PMID: 21120862 Free PMC article.
Cited by
-
Endocannabinoid binding to the cannabinoid receptors: what is known and what remains unknown.Curr Med Chem. 2010;17(14):1468-86. doi: 10.2174/092986710790980005. Curr Med Chem. 2010. PMID: 20166921 Free PMC article. Review.
-
Effect of Temporal Expression of Integral Membrane Proteins by Baculovirus Expression Vector System.Mol Biotechnol. 2018 Aug;60(8):576-584. doi: 10.1007/s12033-018-0099-y. Mol Biotechnol. 2018. PMID: 29943147
-
Molecular dynamics simulation studies of GLUT4: substrate-free and substrate-induced dynamics and ATP-mediated glucose transport inhibition.PLoS One. 2010 Dec 3;5(12):e14217. doi: 10.1371/journal.pone.0014217. PLoS One. 2010. PMID: 21151967 Free PMC article.
-
Understanding functional residues of the cannabinoid CB1.Curr Top Med Chem. 2010;10(8):779-98. doi: 10.2174/156802610791164210. Curr Top Med Chem. 2010. PMID: 20370713 Free PMC article. Review.
-
The cannabinoid type-1 receptor carboxyl-terminus, more than just a tail.J Neurochem. 2011 Apr;117(1):1-18. doi: 10.1111/j.1471-4159.2011.07186.x. Epub 2011 Feb 11. J Neurochem. 2011. PMID: 21244428 Free PMC article. Review.
References
-
- Popot J.L., Engelman D.M. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 2000;69:881–922. - PubMed
-
- Engelman D.M., Chen Y., Chin C.N., Curran A.R., Dixon A.M. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003;555:122–125. - PubMed
-
- Lüneberg J., Widmann M., Dathe M., Marti T. Secondary structure of bacteriorhodopsin fragments. External sequence constraints specify the conformation of transmembrane helices. J. Biol. Chem. 1998;273:28822–28830. - PubMed
-
- Yeagle P.L., Choi G., Albert A.D. Studies on the structure of the G-protein-coupled receptor rhodopsin including the putative G-protein binding site in unactivated and activated forms. Biochemistry. 2001;40:11932–11937. - PubMed
-
- Schneider D. Rendezvous in a membrane: close packing, hydrogen bonding, and the formation of transmembrane helix oligomers. FEBS Lett. 2004;577:5–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
