

Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors
- PMID: 19384205
- PMCID: PMC2743203
- DOI: 10.1097/CCM.0b013e3181a001ae
Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors
Abstract
Objective: Necrotic cells evoke potent innate immune responses through unclear mechanisms. The mitochondrial fraction of the cell retains constituents of its bacterial ancestors, including N-formyl peptides, which are potentially immunogenic. Thus, we hypothesized that the mitochondrial fraction of the cell, particularly N-formyl peptides, contributes significantly to the activation of monocytes by necrotic cells.
Design: Human peripheral blood monocytes were incubated with necrotic cell fractions and mitochondrial proteins to investigate their potential for immune cell activation.
Setting: University Medical Center Research Laboratory.
Subjects: Healthy human adults served as blood donors.
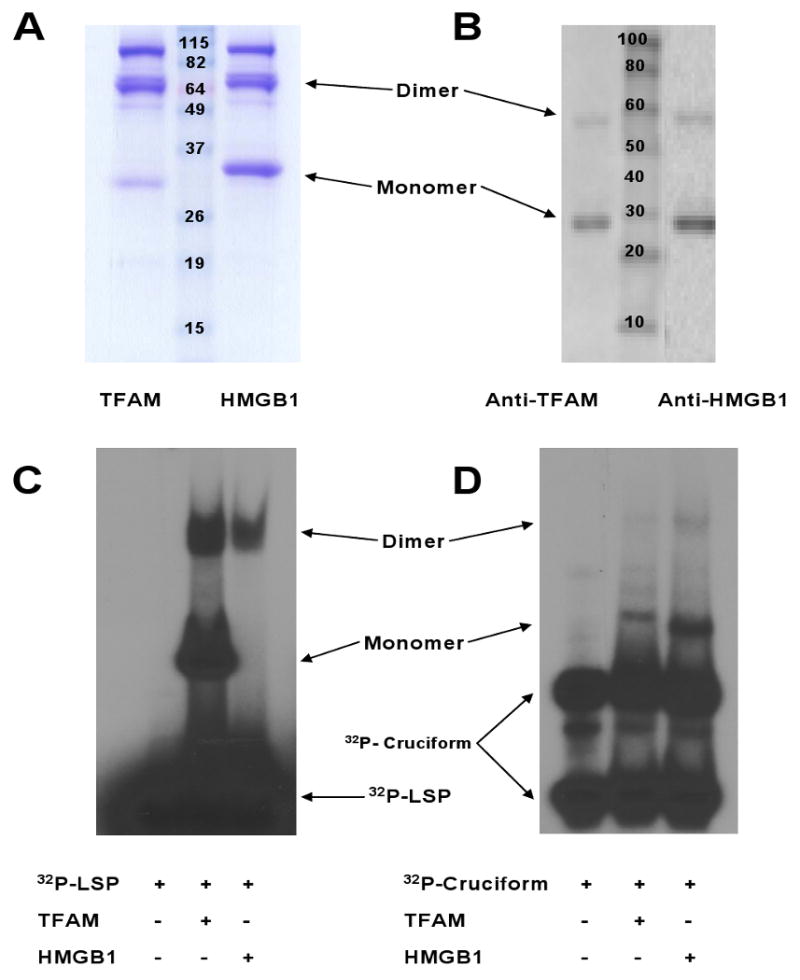
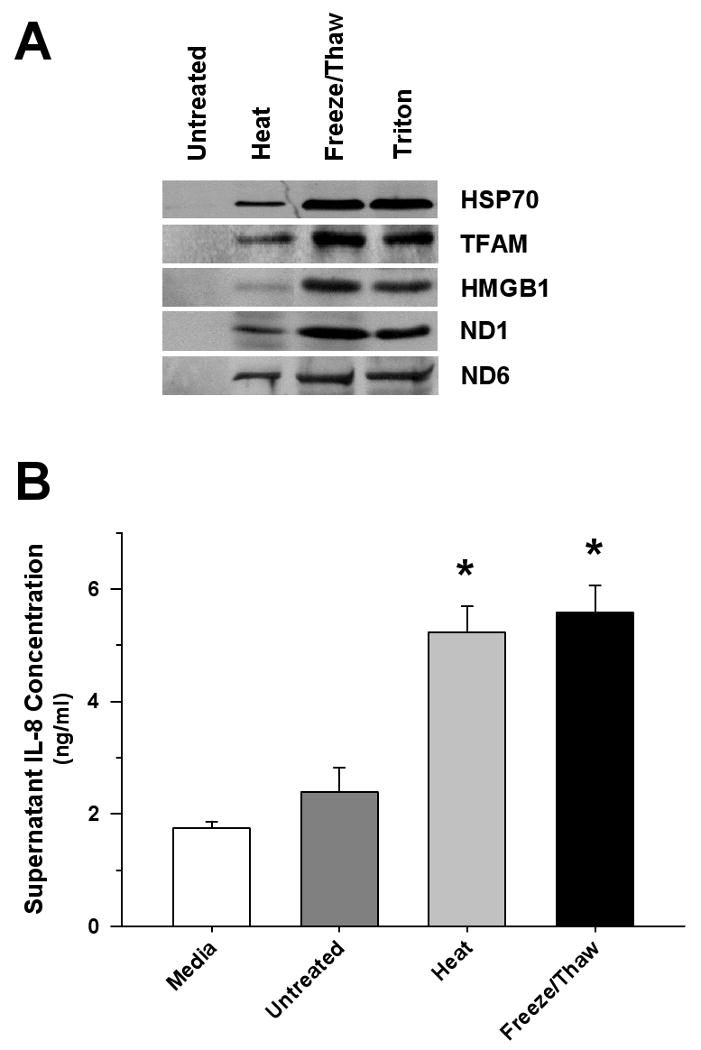
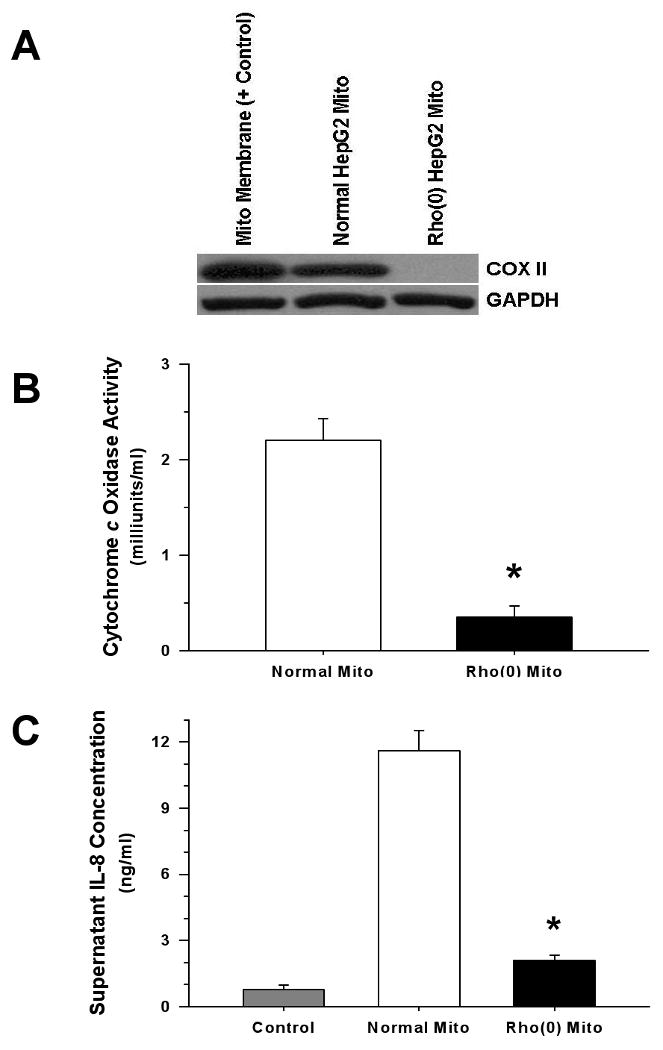
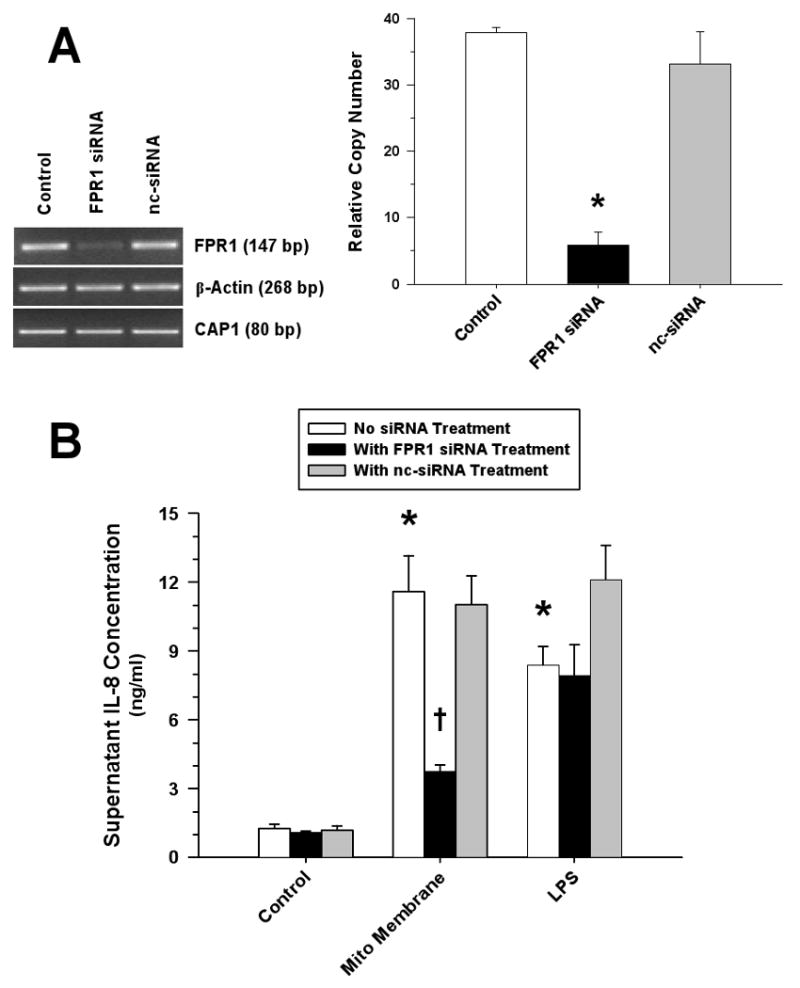
Measurements and main results: Human blood monocyte activation was measured after treatment with cytosolic, nuclear and mitochondrial fractions of necrotic HepG2 cells or necrotic HepG2 cells depleted of N-formyl peptides [Rho(0) cells]. The specific role of the high affinity formyl peptide receptor (FPR) was then tested using specific pharmacologic inhibitors and RNA silencing. The capacity of mitochondrial N-formyl peptides to activate monocytes was confirmed using a synthetic peptide conforming to the N-terminus of mitochondrial nicotinamide adenine dinucleotide subunit 6. The results demonstrated that mitochondrial cell fractions most potently activated monocytes, and interleukin (IL)-8 was selectively released at low-protein concentrations. Mitochondria from Rho(0) cells induced minimal monocyte IL-8 release, and specific pharmacologic inhibitors and RNA-silencing confirmed that FPR contributes significantly to monocyte IL-8 responses to both necrotic cells and mitochondrial proteins. N-formyl peptides alone did not induce monocyte IL-8 release; whereas, the combination of mitochondrial N-formyl peptides and mitochondrial transcription factor A (TFAM) dramatically increased IL-8 release from monocytes. Likewise, high mobility group box 1, the nuclear homolog of TFAM, did not induce monocyte IL-8 release unless combined with mitochondrial N-formyl peptides.
Conclusions: Interactions between mitochondrial N-formyl peptides and FPR in the presence of other mitochondrial antigens (e.g., TFAM) contributes significantly to the activation of monocytes by necrotic cells.
Figures






References
-
- Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. - PubMed
-
- Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. - PubMed
-
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med. 2007;13:851–856. - PubMed
-
- Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75:995–1000. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
