

Identifying native-like protein structures with scoring functions based on all-atom ECEPP force fields, implicit solvent models and structure relaxation
- PMID: 19384995
- PMCID: PMC4502597
- DOI: 10.1002/prot.22414
Identifying native-like protein structures with scoring functions based on all-atom ECEPP force fields, implicit solvent models and structure relaxation
Abstract
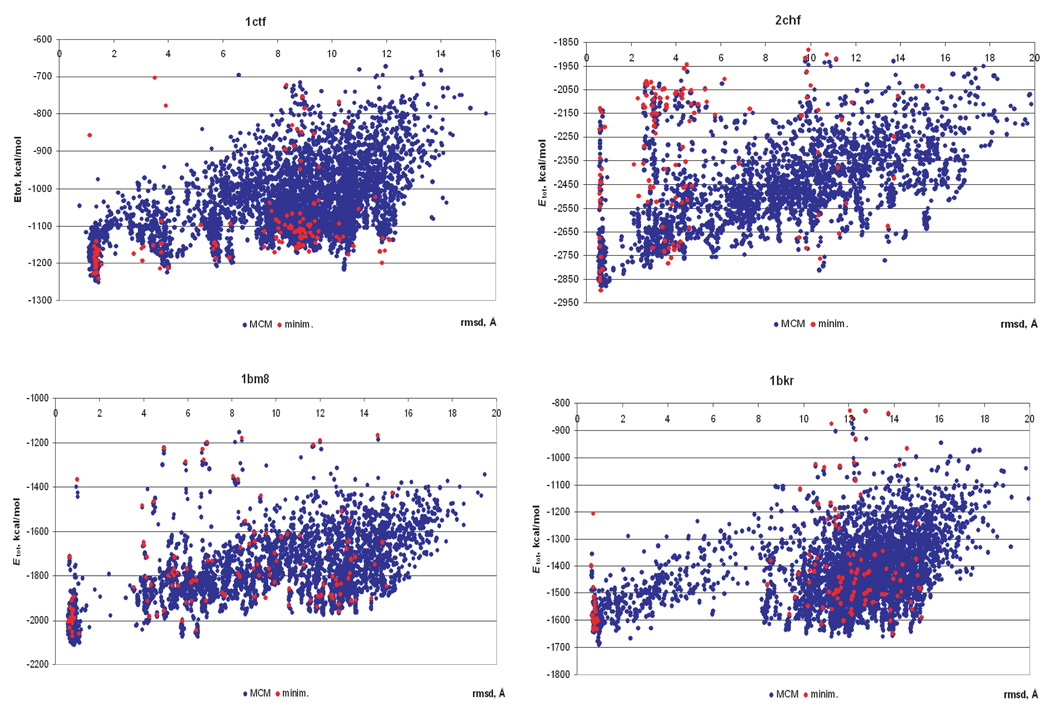
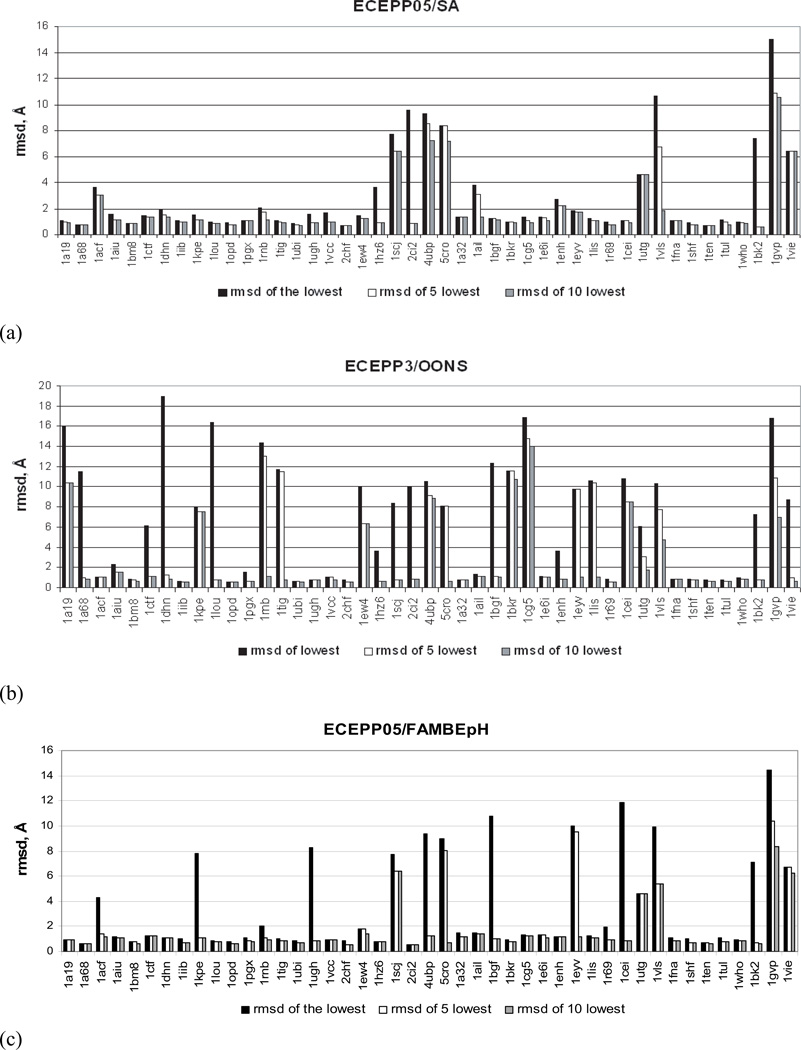
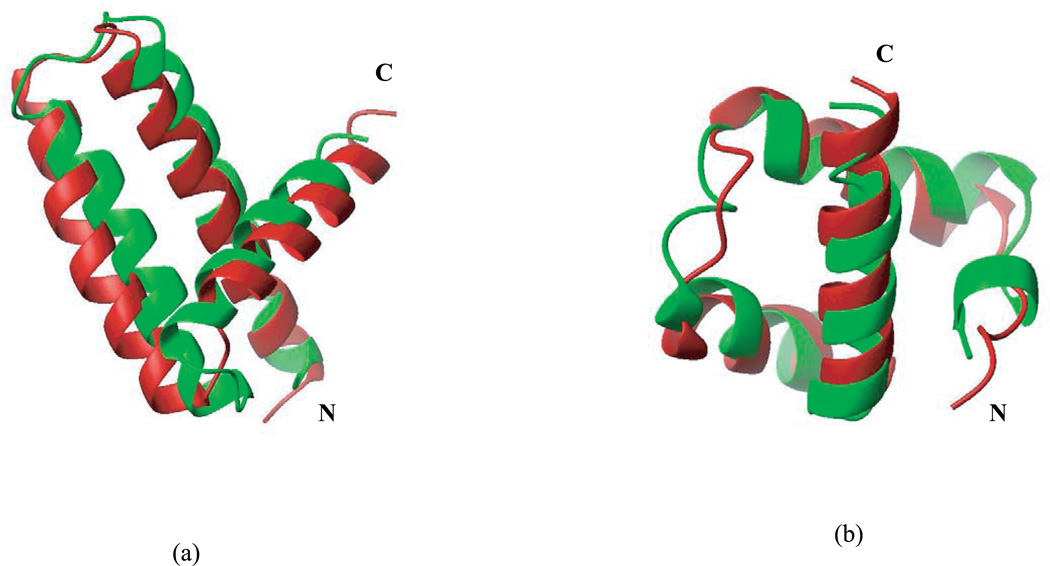
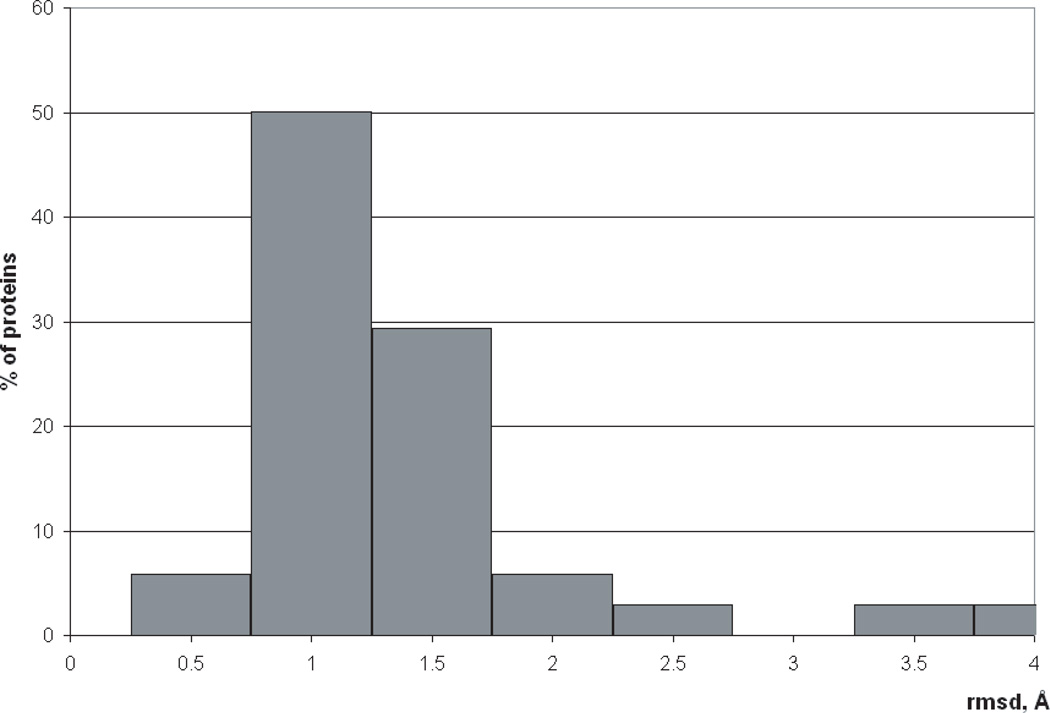
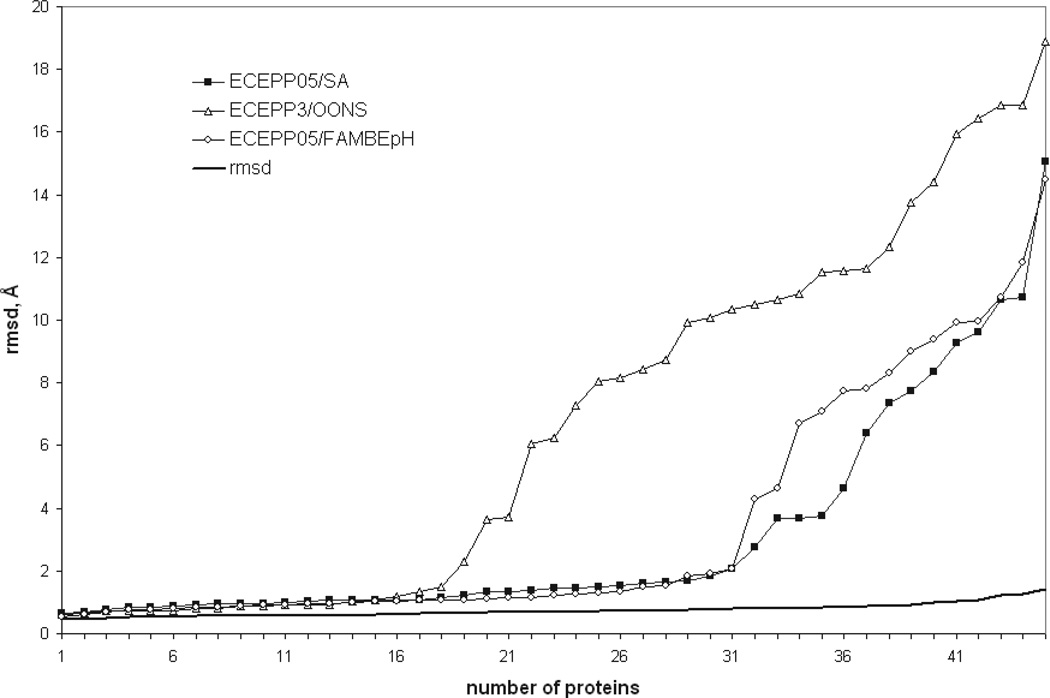
Availability of energy functions which can discriminate native-like from non-native protein conformations is crucial for theoretical protein structure prediction and refinement of low-resolution protein models. This article reports the results of benchmark tests for scoring functions based on two all-atom ECEPP force fields, that is, ECEPP/3 and ECEPP05, and two implicit solvent models for a large set of protein decoys. The following three scoring functions are considered: (i) ECEPP05 plus a solvent-accessible surface area model with the parameters optimized with a set of protein decoys (ECEPP05/SA); (ii) ECEPP/3 plus the solvent-accessible surface area model of Ooi et al. (Proc Natl Acad Sci USA 1987;84:3086-3090) (ECEPP3/OONS); and (iii) ECEPP05 plus an implicit solvent model based on a solution of the Poisson equation with an optimized Fast Adaptive Multigrid Boundary Element (FAMBEpH) method (ECEPP05/FAMBEpH). Short Monte Carlo-with-Minimization (MCM) simulations, following local energy minimization, are used as a scoring method with ECEPP05/SA and ECEPP3/OONS potentials, whereas energy calculation is used with ECEPP05/FAMBEpH. The performance of each scoring function is evaluated by examining its ability to distinguish between native-like and non-native protein structures. The results of the tests show that the new ECEPP05/SA scoring function represents a significant improvement over the earlier ECEPP3/OONS version of the force field. Thus, it is able to rank native-like structures with C(alpha) root-mean-square-deviations below 3.5 A as lowest-energy conformations for 76% and within the top 10 for 87% of the proteins tested, compared with 69 and 80%, respectively, for ECEPP3/OONS. The use of the FAMBEpH solvation model, which provides a more accurate description of the protein-solvent interactions, improves the discriminative ability of the scoring function to 89%. All failed tests in which the native-like structures cannot be discriminated as those with low energy, are due to omission of protein-protein interactions. The results of this study represent a benchmark in force-field development, and may be useful for evaluation of the performance of different force fields.
Figures
Similar articles
-
Use of decoys to optimize an all-atom force field including hydration.Biophys J. 2008 Sep;95(5):2434-49. doi: 10.1529/biophysj.108.133587. Epub 2008 May 23. Biophys J. 2008. PMID: 18502794 Free PMC article.
-
Physical scoring function based on AMBER force field and Poisson-Boltzmann implicit solvent for protein structure prediction.Proteins. 2004 Aug 15;56(3):475-86. doi: 10.1002/prot.20133. Proteins. 2004. PMID: 15229881
-
Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the OPLS all-atom force field and the Surface Generalized Born solvent model.Proteins. 2002 Aug 1;48(2):404-22. doi: 10.1002/prot.10171. Proteins. 2002. PMID: 12112706
-
Methods for the Refinement of Protein Structure 3D Models.Int J Mol Sci. 2019 May 9;20(9):2301. doi: 10.3390/ijms20092301. Int J Mol Sci. 2019. PMID: 31075942 Free PMC article. Review.
-
Advances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins at constant pH.Adv Protein Chem Struct Biol. 2011;85:281-322. doi: 10.1016/B978-0-12-386485-7.00008-9. Adv Protein Chem Struct Biol. 2011. PMID: 21920327 Review.
Cited by
-
Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity.Viruses. 2023 Jan 22;15(2):309. doi: 10.3390/v15020309. Viruses. 2023. PMID: 36851526 Free PMC article.
-
Coupled molecular dynamics and continuum electrostatic method to compute the ionization pKa's of proteins as a function of pH. Test on a large set of proteins.J Biomol Struct Dyn. 2018 Feb;36(3):561-574. doi: 10.1080/07391102.2017.1288169. Epub 2017 Feb 24. J Biomol Struct Dyn. 2018. PMID: 28132613 Free PMC article.
-
Further evidence for the likely completeness of the library of solved single domain protein structures.J Phys Chem B. 2012 Jun 14;116(23):6654-64. doi: 10.1021/jp211052j. Epub 2012 Feb 13. J Phys Chem B. 2012. PMID: 22272723 Free PMC article.
-
Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2.Comput Struct Biotechnol J. 2022;20:2091-2111. doi: 10.1016/j.csbj.2022.04.010. Epub 2022 Apr 9. Comput Struct Biotechnol J. 2022. PMID: 35432786 Free PMC article.
-
Development of a new physics-based internal coordinate mechanics force field and its application to protein loop modeling.Proteins. 2011 Feb;79(2):477-98. doi: 10.1002/prot.22896. Proteins. 2011. PMID: 21069716 Free PMC article.
References
-
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. - PubMed
-
- Das R, Qian B, Raman S, Vernon R, Thompson J, Bradley P, Khare S, Tyka MD, Bhat D, Chivian D, Kim DE, Sheffler WH, Malmström L, Wollacott AM, Wang C, Andre I, Baker D. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@home. Proteins: Struct Funct Bioinform. 2007;69(Suppl 8):118–128. - PubMed
-
- Fisher D. 3D–SHOTGUN: a novel, cooperative, fold-recognition meta-predictor. Proteins. 2003;51:434–444. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
