

Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower
- PMID: 19385720
- PMCID: PMC2671559
- DOI: 10.1371/journal.pbio.1000090
Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower
Abstract
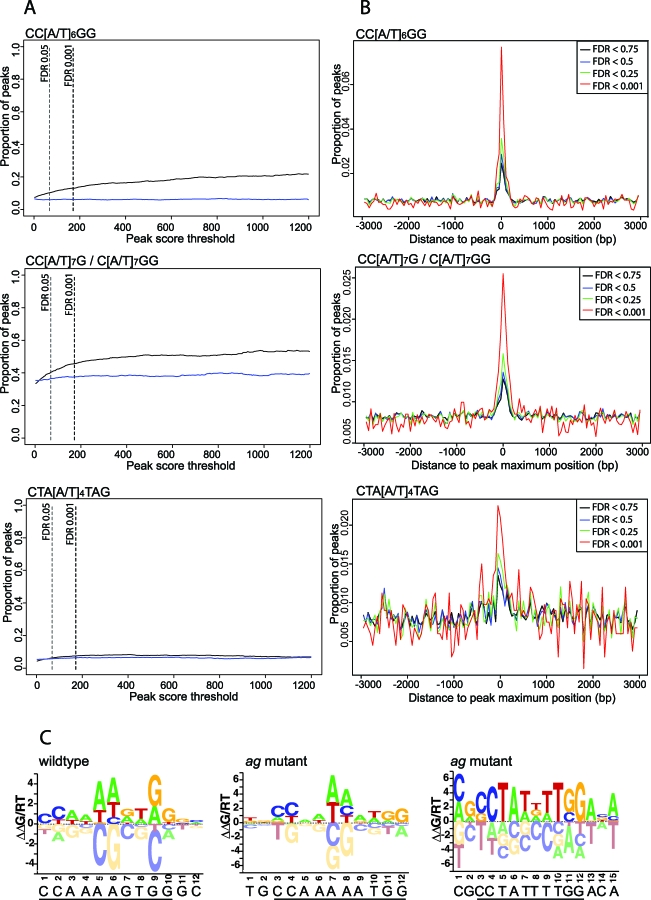
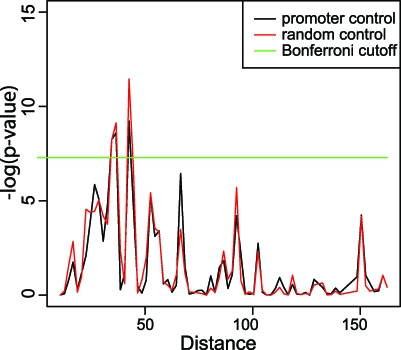
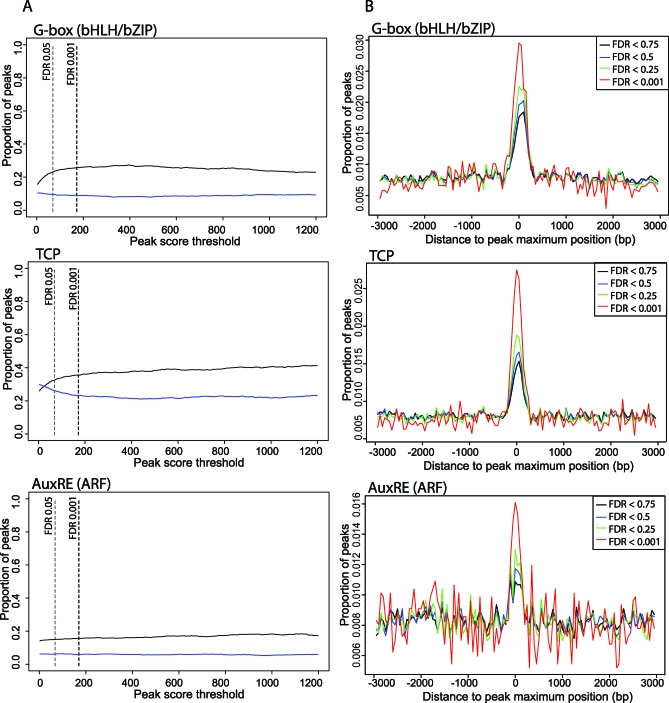
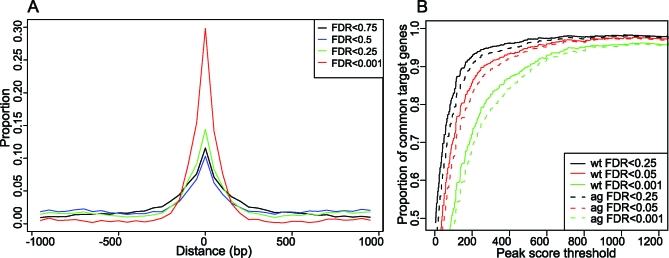
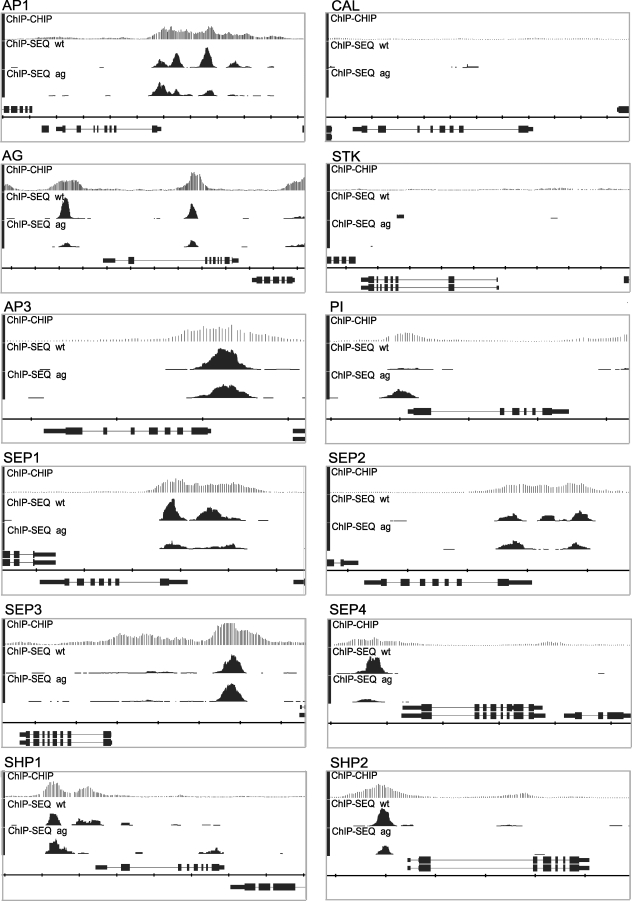
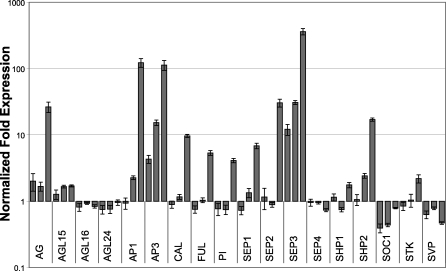
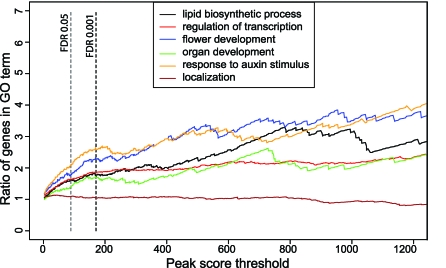
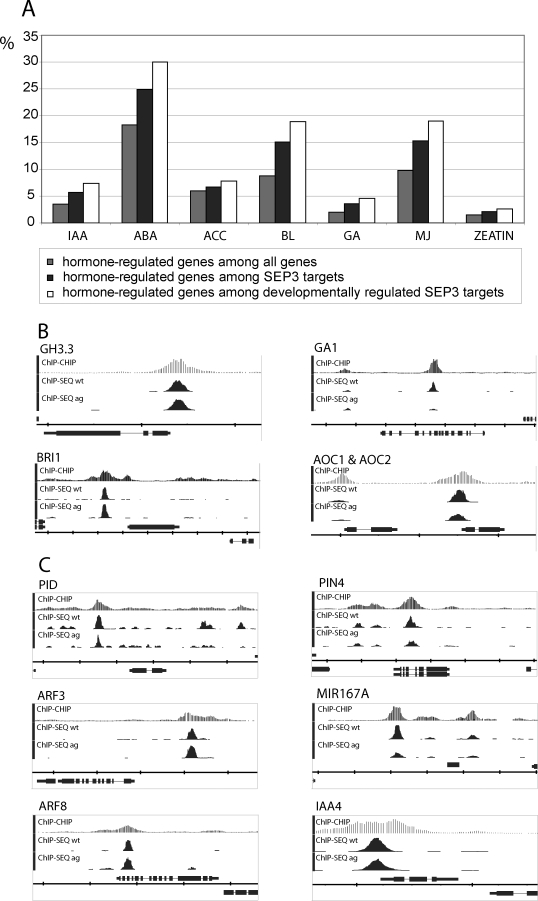
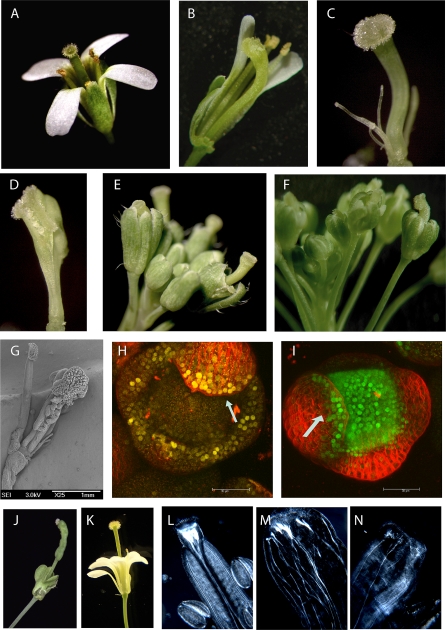
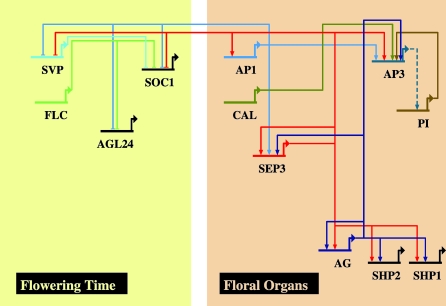
The molecular mechanisms by which floral homeotic genes act as major developmental switches to specify the identity of floral organs are still largely unknown. Floral homeotic genes encode transcription factors of the MADS-box family, which are supposed to assemble in a combinatorial fashion into organ-specific multimeric protein complexes. Major mediators of protein interactions are MADS-domain proteins of the SEPALLATA subfamily, which play a crucial role in the development of all types of floral organs. In order to characterize the roles of the SEPALLATA3 transcription factor complexes at the molecular level, we analyzed genome-wide the direct targets of SEPALLATA3. We used chromatin immunoprecipitation followed by ultrahigh-throughput sequencing or hybridization to whole-genome tiling arrays to obtain genome-wide DNA-binding patterns of SEPALLATA3. The results demonstrate that SEPALLATA3 binds to thousands of sites in the genome. Most potential target sites that were strongly bound in wild-type inflorescences are also bound in the floral homeotic agamous mutant, which displays only the perianth organs, sepals, and petals. Characterization of the target genes shows that SEPALLATA3 integrates and modulates different growth-related and hormonal pathways in a combinatorial fashion with other MADS-box proteins and possibly with non-MADS transcription factors. In particular, the results suggest multiple links between SEPALLATA3 and auxin signaling pathways. Our gene expression analyses link the genomic binding site data with the phenotype of plants expressing a dominant repressor version of SEPALLATA3, suggesting that it modulates auxin response to facilitate floral organ outgrowth and morphogenesis. Furthermore, the binding of the SEPALLATA3 protein to cis-regulatory elements of other MADS-box genes and expression analyses reveal that this protein is a key component in the regulatory transcriptional network underlying the formation of floral organs.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures
Similar articles
-
Dynamics of chromatin accessibility and gene regulation by MADS-domain transcription factors in flower development.Genome Biol. 2014 Mar 3;15(3):R41. doi: 10.1186/gb-2014-15-3-r41. Genome Biol. 2014. PMID: 24581456 Free PMC article.
-
The floral homeotic protein SEPALLATA3 recognizes target DNA sequences by shape readout involving a conserved arginine residue in the MADS-domain.Plant J. 2018 Jul;95(2):341-357. doi: 10.1111/tpj.13954. Epub 2018 Jun 4. Plant J. 2018. PMID: 29744943
-
DNA-binding properties of the MADS-domain transcription factor SEPALLATA3 and mutant variants characterized by SELEX-seq.Plant Mol Biol. 2021 Mar;105(4-5):543-557. doi: 10.1007/s11103-020-01108-6. Epub 2021 Jan 24. Plant Mol Biol. 2021. PMID: 33486697 Free PMC article.
-
Molecular mechanisms of floral organ specification by MADS domain proteins.Curr Opin Plant Biol. 2016 Feb;29:154-62. doi: 10.1016/j.pbi.2015.12.004. Epub 2016 Jan 21. Curr Opin Plant Biol. 2016. PMID: 26802807 Review.
-
Control of patterning, growth, and differentiation by floral organ identity genes.J Exp Bot. 2015 Feb;66(4):1065-73. doi: 10.1093/jxb/eru514. Epub 2015 Jan 21. J Exp Bot. 2015. PMID: 25609826 Review.
Cited by
-
The MADS Symphonies of Transcriptional Regulation.Front Plant Sci. 2011 Jul 1;2:26. doi: 10.3389/fpls.2011.00026. eCollection 2011. Front Plant Sci. 2011. PMID: 22645529 Free PMC article. No abstract available.
-
Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis.Plant Cell. 2012 Jul;24(7):2719-31. doi: 10.1105/tpc.112.098061. Epub 2012 Jul 5. Plant Cell. 2012. PMID: 22773751 Free PMC article.
-
Identification of MADS-Box Transcription Factors in Iris laevigata and Functional Assessment of IlSEP3 and IlSVP during Flowering.Int J Mol Sci. 2022 Sep 1;23(17):9950. doi: 10.3390/ijms23179950. Int J Mol Sci. 2022. PMID: 36077350 Free PMC article.
-
Arabidopsis AGAMOUS-LIKE16 and SUPPRESSOR OF CONSTANS1 regulate the genome-wide expression and flowering time.Plant Physiol. 2023 May 2;192(1):154-169. doi: 10.1093/plphys/kiad058. Plant Physiol. 2023. PMID: 36721922 Free PMC article.
-
Digital gene expression signatures for maize development.Plant Physiol. 2010 Nov;154(3):1024-39. doi: 10.1104/pp.110.159673. Epub 2010 Sep 10. Plant Physiol. 2010. PMID: 20833728 Free PMC article.
References
-
- Coen E. Goethe and the ABC model of flower development. C R Acad Sci III. 2001;324:523–530. - PubMed
-
- Chuck G, Hake S. Regulation of developmental transitions. Curr Opin Plant Biol. 2005;8:67–70. - PubMed
-
- Theissen G, Becker A, Di Rosa A, Kanno A, Kim JT, et al. A short history of MADS-box genes in plants. Plant Mol Biol. 2000;42:115–149. - PubMed
-
- Ferrario S, Immink RG, Angenent GC. Conservation and diversity in flower land. Curr Opin Plant Biol. 2004;7:84–91. - PubMed
-
- International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
