

KIRMES: kernel-based identification of regulatory modules in euchromatic sequences
- PMID: 19389732
- PMCID: PMC2722996
- DOI: 10.1093/bioinformatics/btp278
KIRMES: kernel-based identification of regulatory modules in euchromatic sequences
Abstract
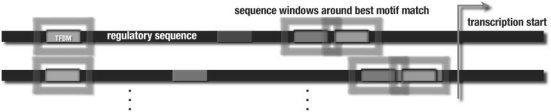
Motivation: Understanding transcriptional regulation is one of the main challenges in computational biology. An important problem is the identification of transcription factor (TF) binding sites in promoter regions of potential TF target genes. It is typically approached by position weight matrix-based motif identification algorithms using Gibbs sampling, or heuristics to extend seed oligos. Such algorithms succeed in identifying single, relatively well-conserved binding sites, but tend to fail when it comes to the identification of combinations of several degenerate binding sites, as those often found in cis-regulatory modules.
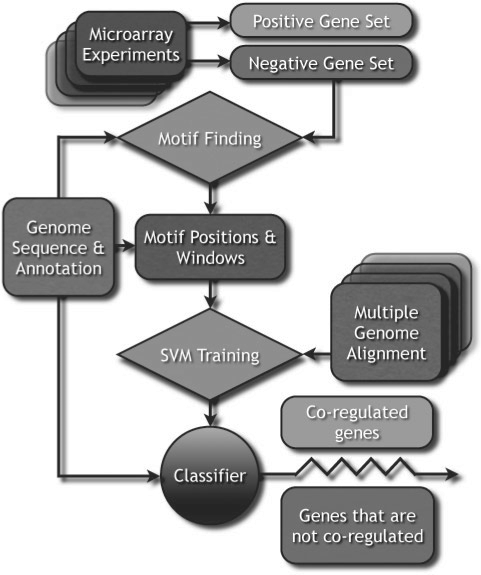
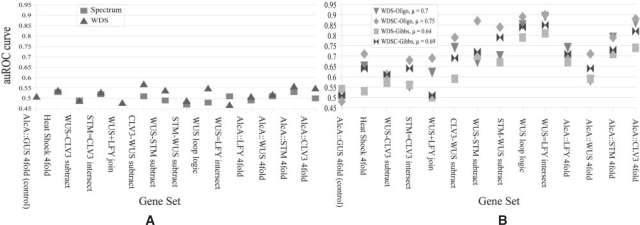
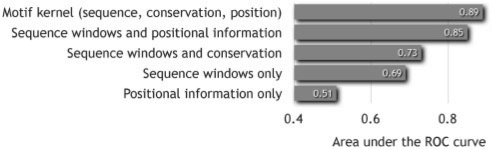
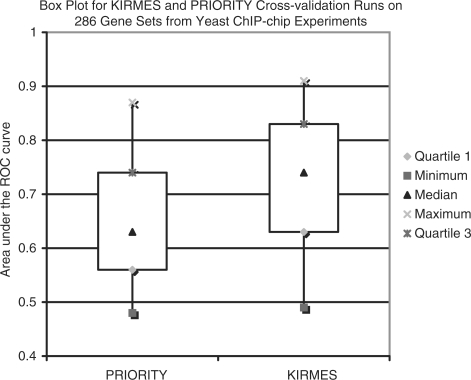
Results: We propose a new algorithm that combines the benefits of existing motif finding with the ones of support vector machines (SVMs) to find degenerate motifs in order to improve the modeling of regulatory modules. In experiments on microarray data from Arabidopsis thaliana, we were able to show that the newly developed strategy significantly improves the recognition of TF targets.
Availability: The python source code (open source-licensed under GPL), the data for the experiments and a Galaxy-based web service are available at http://www.fml.mpg.de/raetsch/suppl/kirmes/.
Figures
References
-
- Bailey T, Elkan C. Proceedings of ISMB'94. Vol. 2. Menlo Park, CA, USA, ISCB: AAAI Press; 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers; pp. 28–36. - PubMed
-
- Boser B, et al. Proceedings COLT '92. Pittsburgh, Pennsylvania: ACM Press; 1992. A training algorithm for optimal margin classifiers; pp. 144–152.
-
- Busch W, et al. Identification of novel heat shock factor-dependent genes and biochemical pathways in A. thaliana. Plant J. 2005;41:1–14. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
