

Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation
- PMID: 19390091
- PMCID: PMC2675865
- DOI: 10.1101/gad.1773909
Bile acid signaling pathways increase stability of Small Heterodimer Partner (SHP) by inhibiting ubiquitin-proteasomal degradation
Abstract
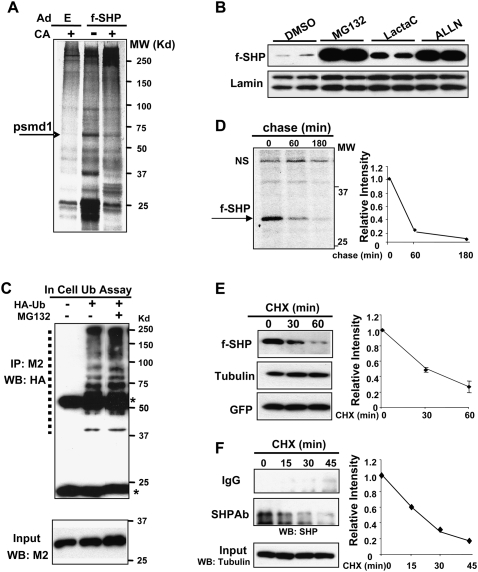
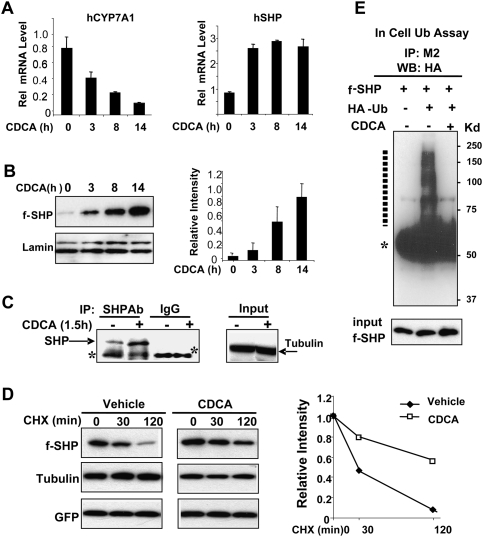
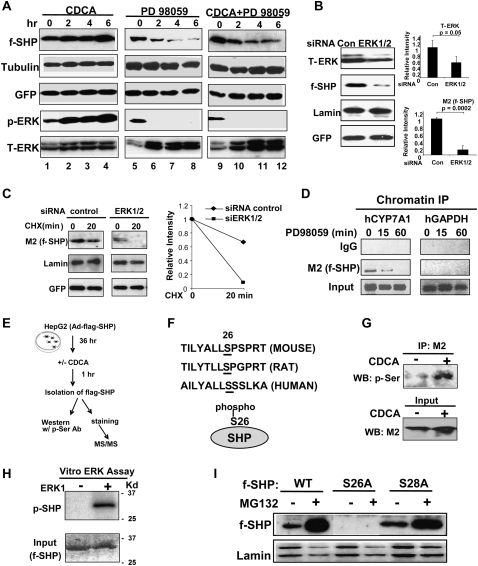
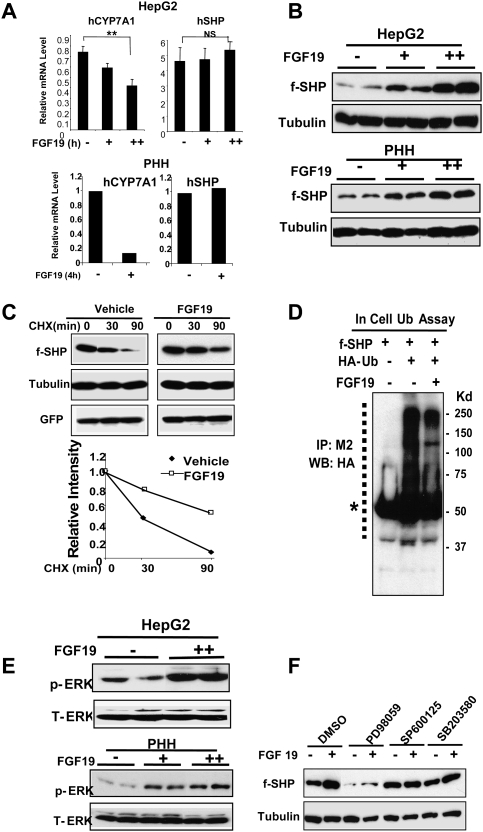
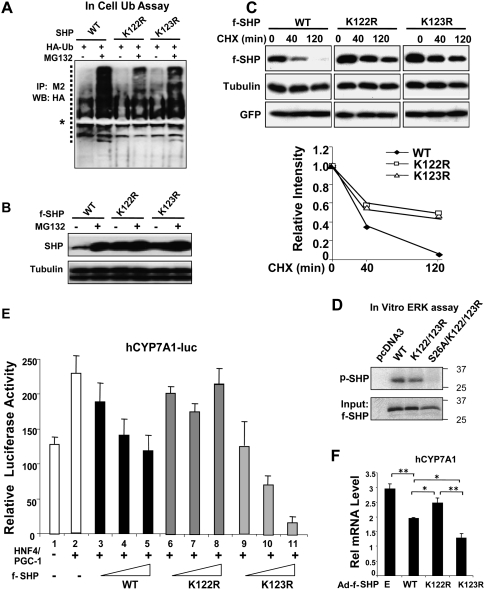
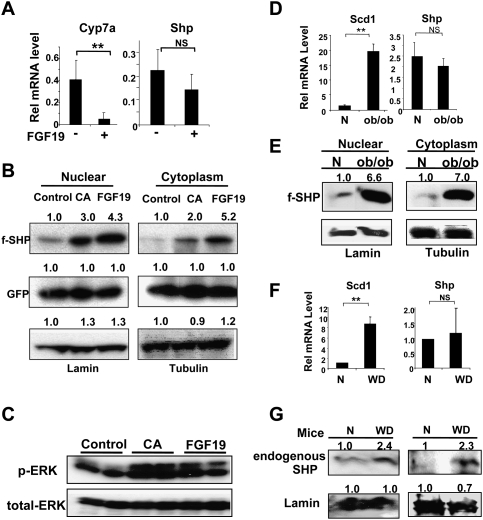
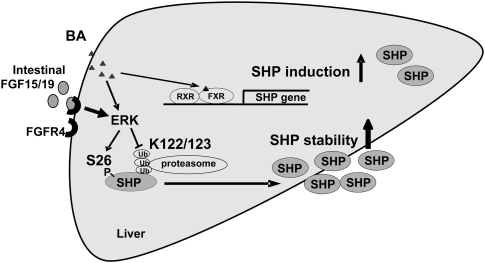
Small Heterodimer Partner (SHP) inhibits activities of numerous transcription factors involved in diverse biological pathways. As an important metabolic regulator, SHP plays a key role in maintaining cholesterol and bile acid homeostasis by inhibiting cholesterol conversion to bile acids. While SHP gene induction by increased bile acids is well established, whether SHP activity is also modulated remains unknown. Here, we report surprising findings that SHP is a rapidly degraded protein via the ubiquitin-proteasomal pathway and that bile acids or bile acid-induced intestinal fibroblast growth factor 19 (FGF19) increases stability of hepatic SHP by inhibiting proteasomal degradation in an extracellular signal-regulated kinase (ERK)-dependent manner. SHP was ubiquitinated at Lys122 and Lys123, and mutation of these sites altered its stability and repression activity. Tandem mass spectrometry revealed that upon bile acid treatment, SHP was phosphorylated at Ser26, within an ERK motif in SHP, and mutation of this site dramatically abolished SHP stability. Surprisingly, SHP stability was abnormally elevated in ob/ob mice and diet-induced obese mice. These results demonstrate an important role for regulation of SHP stability in bile acid signaling in normal conditions, and that abnormal stabilization of SHP may be associated with metabolic disorders, including obesity and diabetes.
Figures
References
-
- Bavner A., Sanyal S., Gustafsson J.A., Treuter E. Transcriptional corepression by SHP: Molecular mechanisms and physiological consequences. Trends Endocrinol. Metab. 2005;16:478–488. - PubMed
-
- Brooks C.L., Gu W. Ubiquitination, phosphorylation and acetylation: The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 2003;15:164–171. - PubMed
-
- Chiang J.Y.L. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002;23:443–463. - PubMed
-
- Deng S., Zhou H., Xiong R., Lu Y., Yan D., Xing T., Dong L., Tang E., Yang H. Over-expression of genes and proteins of ubiquitin specific peptidases (USPs) and proteasome subunits (PSs) in breast cancer tissue observed by the methods of RFDD-PCR and proteomics. Breast Cancer Res. Treat. 2007;104:21–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous