

Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses
- PMID: 19390600
- PMCID: PMC2666264
- DOI: 10.1371/journal.ppat.1000397
Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses
Abstract
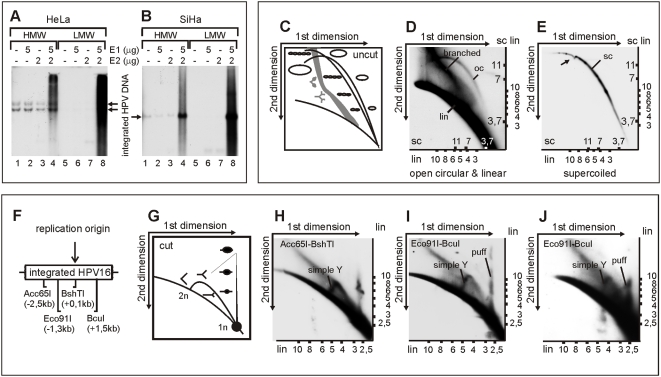
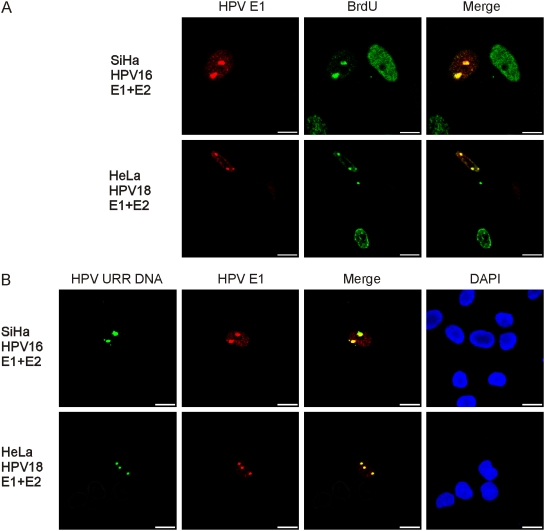
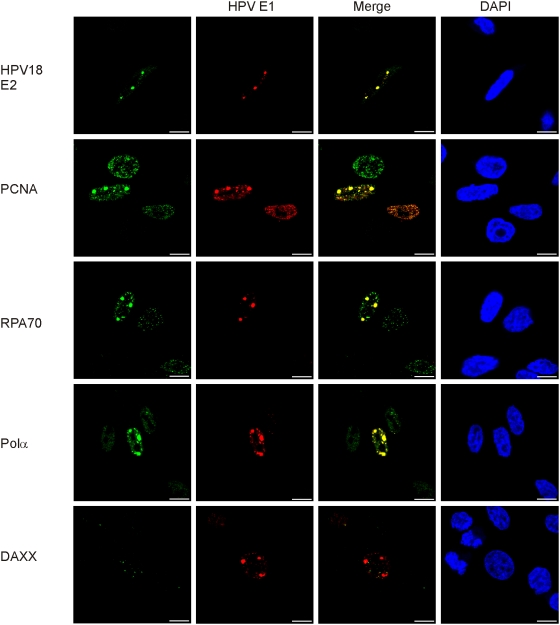
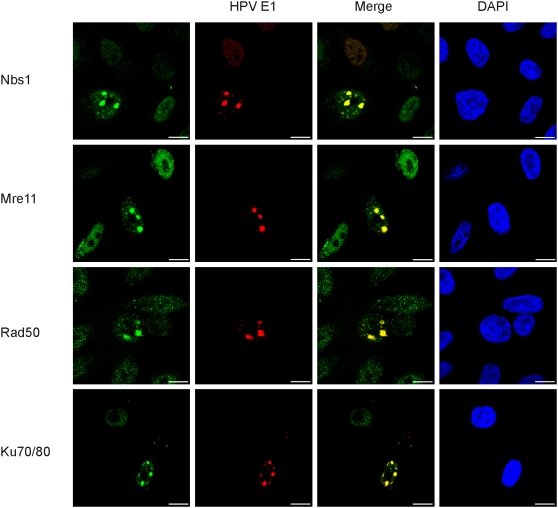
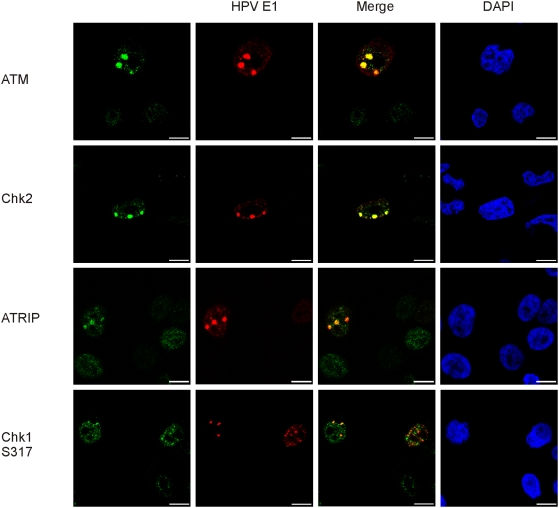
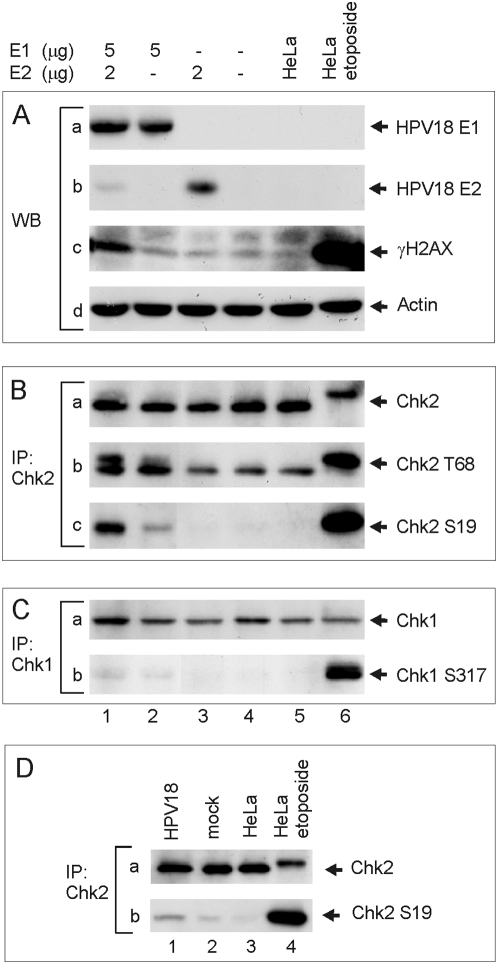
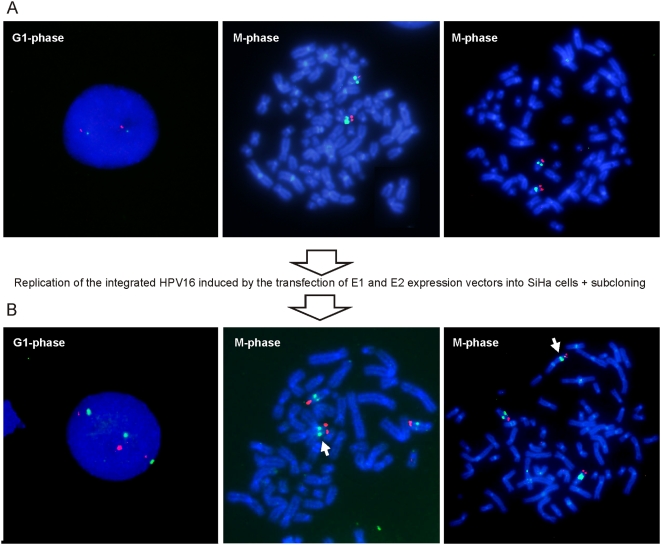
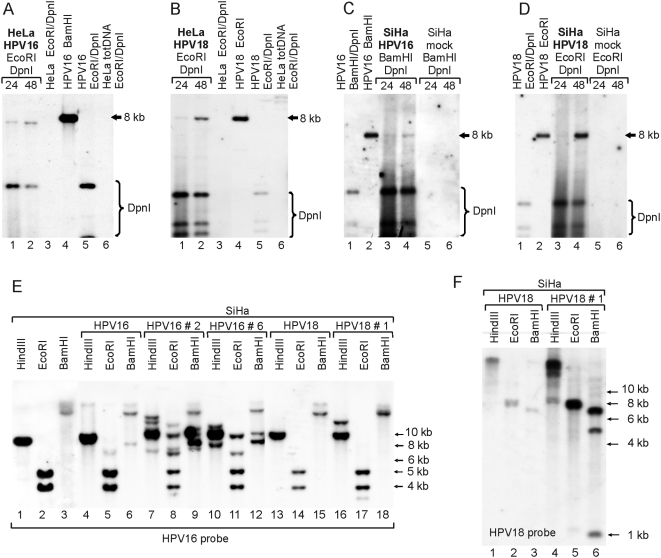
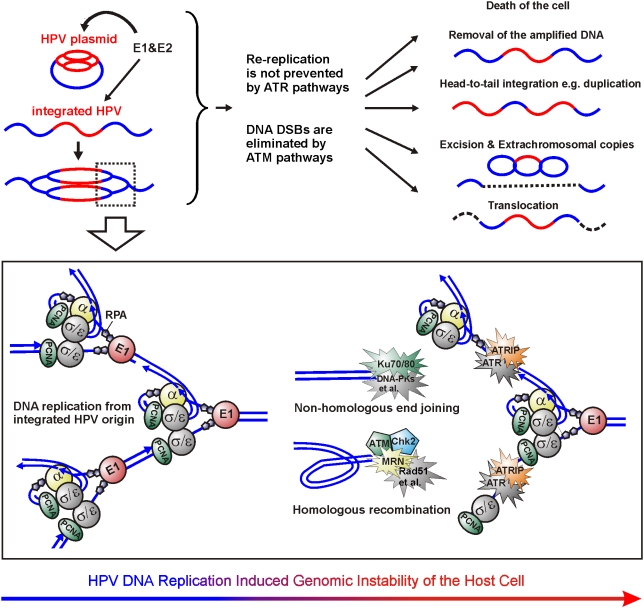
In HPV-related cancers, the "high-risk" human papillomaviruses (HPVs) are frequently found integrated into the cellular genome. The integrated subgenomic HPV fragments express viral oncoproteins and carry an origin of DNA replication that is capable of initiating bidirectional DNA re-replication in the presence of HPV replication proteins E1 and E2, which ultimately leads to rearrangements within the locus of the integrated viral DNA. The current study indicates that the E1- and E2-dependent DNA replication from the integrated HPV origin follows the "onion skin"-type replication mode and generates a heterogeneous population of replication intermediates. These include linear, branched, open circular, and supercoiled plasmids, as identified by two-dimensional neutral-neutral gel-electrophoresis. We used immunofluorescence analysis to show that the DNA repair/recombination centers are assembled at the sites of the integrated HPV replication. These centers recruit viral and cellular replication proteins, the MRE complex, Ku70/80, ATM, Chk2, and, to some extent, ATRIP and Chk1 (S317). In addition, the synthesis of histone gammaH2AX, which is a hallmark of DNA double strand breaks, is induced, and Chk2 is activated by phosphorylation in the HPV-replicating cells. These changes suggest that the integrated HPV replication intermediates are processed by the activated cellular DNA repair/recombination machinery, which results in cross-chromosomal translocations as detected by metaphase FISH. We also confirmed that the replicating HPV episomes that expressed the physiological levels of viral replication proteins could induce genomic instability in the cells with integrated HPV. We conclude that the HPV replication origin within the host chromosome is one of the key factors that triggers the development of HPV-associated cancers. It could be used as a starting point for the "onion skin"-type of DNA replication whenever the HPV plasmid exists in the same cell, which endangers the host genomic integrity during the initial integration and after the de novo infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Howley PM, Lowy DR. Papillomaviruses and Their Replication. In: Knipe DM, aH PM, editors. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 2197–2230.
-
- Kadaja M, Silla T, Ustav E, Ustav M. Papillomavirus DNA replication - from initiation to genomic instability. Virology. 2009;384:360–368. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
