

Autosomal recessive cutis laxa syndrome revisited
- PMID: 19401719
- PMCID: PMC2986595
- DOI: 10.1038/ejhg.2009.22
Autosomal recessive cutis laxa syndrome revisited
Abstract
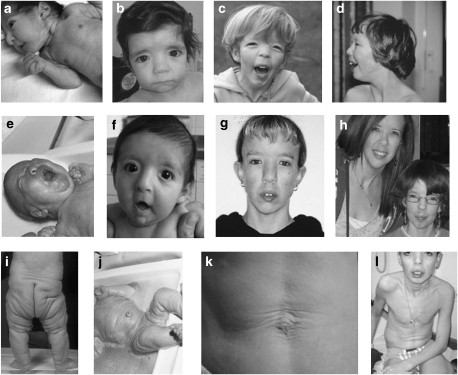
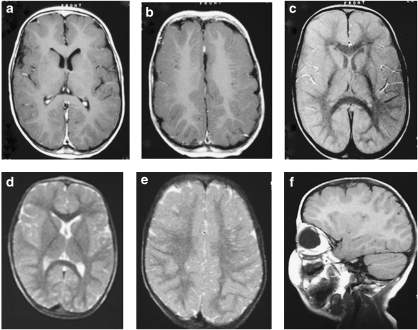
The clinical spectrum of the autosomal recessive cutis laxa syndromes is highly heterogeneous with respect to organ involvement and severity. One of the major diagnostic criteria is to detect abnormal elastin fibers. In several other clinically similar autosomal recessive syndromes, however, the classic histological anomalies are absent, and the definite diagnosis remains uncertain. In cutis laxa patients mutations have been demonstrated in elastin or fibulin genes, but in the majority of patients the underlying genetic etiology remains unknown. Recently, we found mutations in the ATP6V0A2 gene in families with autosomal recessive cutis laxa. This genetic defect is associated with abnormal glycosylation leading to a distinct combined disorder of the biosynthesis of N- and O-linked glycans. Interestingly, similar mutations have been found in patients with wrinkly skin syndrome, without the presence of severe skin symptoms of elastin deficiency. These findings suggest that the cutis laxa and wrinkly skin syndromes are phenotypic variants of the same disorder. Interestingly many phenotypically similar patients carry no mutations in the ATP6V0A2 gene. The variable presence of protein glycosylation abnormalities in the diverse clinical forms of the wrinkled skin-cutis laxa syndrome spectrum necessitates revisiting the diagnostic criteria to be able to offer adequate prognosis assessment and counseling. This paper aims at describing the spectrum of clinical features of the various forms of autosomal recessive cutis laxa syndromes. Based on the recently unraveled novel genetic entity we also review the genetic aspects in cutis laxa syndromes including genotype-phenotype correlations and suggest a practical diagnostic approach.
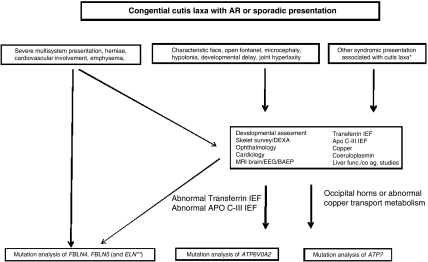
Figures
Comment in
-
Lessons from cutis laxa syndromes: wrinkles due to improper reloading of the extracellular matrix?Eur J Hum Genet. 2009 Sep;17(9):1097-8. doi: 10.1038/ejhg.2009.59. Epub 2009 Apr 29. Eur J Hum Genet. 2009. PMID: 19401717 Free PMC article. No abstract available.
-
De Barsy syndrome and ATP6V0A2-CDG.Eur J Hum Genet. 2010 May;18(5):526; author reply 526. doi: 10.1038/ejhg.2009.218. Epub 2009 Dec 16. Eur J Hum Genet. 2010. PMID: 20010974 Free PMC article. No abstract available.
References
-
- Urban Z, Gao J, Pope FM, Davis EC. Autosomal dominant cutis laxa with severe lung disease: synthesis and matrix deposition of mutant tropoelastin. J Invest Dermatol. 2005;124:1193–1199. - PubMed
-
- Morava E, Lefeber DJ, Urban Z, et al. Defining the phenotype in an autosomal recessive cutis laxa syndrome with a combined congenital defect of glycosylation. Eur J Hum Genet. 2008;16:28–35. - PubMed
-
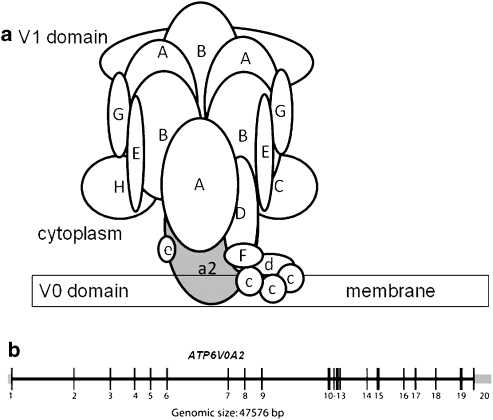
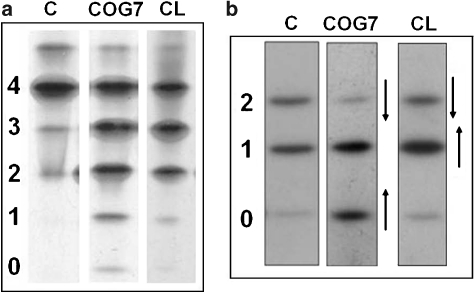
- Kornak U, Reynders E, Dimopoulou A, et al. Impaired glycosylation and cutis laxa caused by mutations in the vesicular H+-ATPase subunit ATP6V0A2. Nat Genet. 2008;40:32–34. - PubMed
-
- Morava E, Wopereis S, Coucke P, et al. Defective protein glycosylation in patients with cutis laxa syndrome. Eur J Hum Genet. 2005;13:414–421. - PubMed
-
- Mehregan AH, Lee SC, Nabai H: Cutis laxa (generalized elastolysis) A report of four cases with autopsy findings. J Cut Path. 1978;5:116–126. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
