

Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons
- PMID: 19404401
- PMCID: PMC2671839
- DOI: 10.1371/journal.pone.0005390
Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons
Abstract
Background: Amyotrophic lateral sclerosis (ALS), the most frequent adult onset motor neuron disease, is associated with hypermetabolism linked to defects in muscle mitochondrial energy metabolism such as ATP depletion and increased oxygen consumption. It remains unknown whether muscle abnormalities in energy metabolism are causally involved in the destruction of neuromuscular junction (NMJ) and subsequent motor neuron degeneration during ALS.
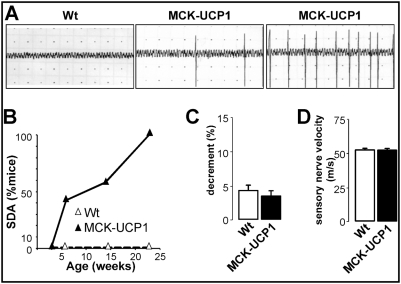
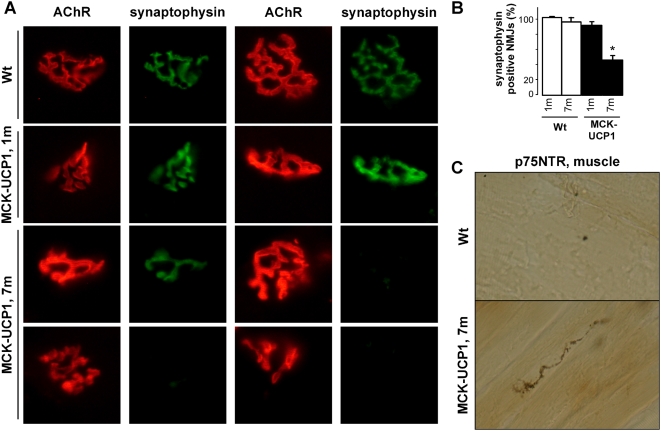
Methodology/principal findings: We studied transgenic mice with muscular overexpression of uncoupling protein 1 (UCP1), a potent mitochondrial uncoupler, as a model of muscle restricted hypermetabolism. These animals displayed age-dependent deterioration of the NMJ that correlated with progressive signs of denervation and a mild late-onset motor neuron pathology. NMJ regeneration and functional recovery were profoundly delayed following injury of the sciatic nerve and muscle mitochondrial uncoupling exacerbated the pathology of an ALS animal model.
Conclusions/significance: These findings provide the proof of principle that a muscle restricted mitochondrial defect is sufficient to generate motor neuron degeneration and suggest that therapeutic strategies targeted at muscle metabolism might prove useful for motor neuron diseases.
Conflict of interest statement
Figures







References
-
- Gonzalez de Aguilar JL, Echaniz-Laguna A, Fergani A, Rene F, Meininger V, et al. Amyotrophic lateral sclerosis: all roads lead to Rome. J Neurochem 2007 - PubMed
-
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. - PubMed
-
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. - PubMed
-
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. - PubMed
-
- Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, et al. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. 2001;74:328–334. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
