

Light sheet-based fluorescence microscopy: more dimensions, more photons, and less photodamage
- PMID: 19404438
- PMCID: PMC2639947
- DOI: 10.2976/1.2974980
Light sheet-based fluorescence microscopy: more dimensions, more photons, and less photodamage
Abstract
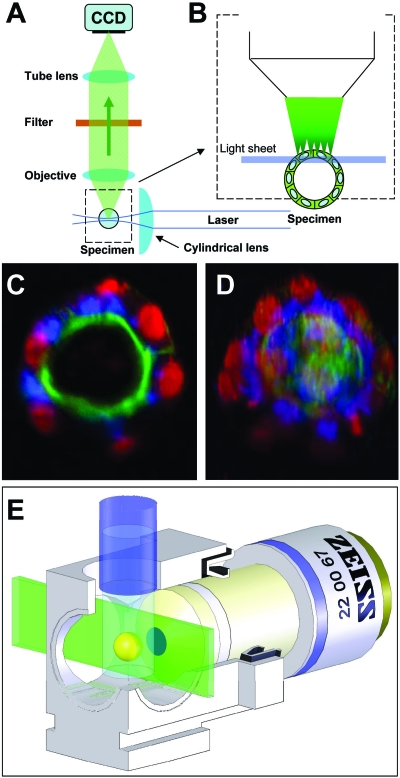
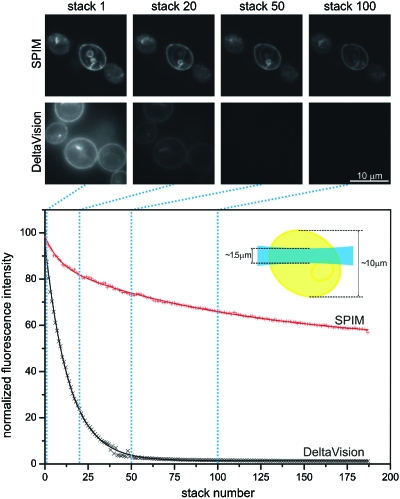
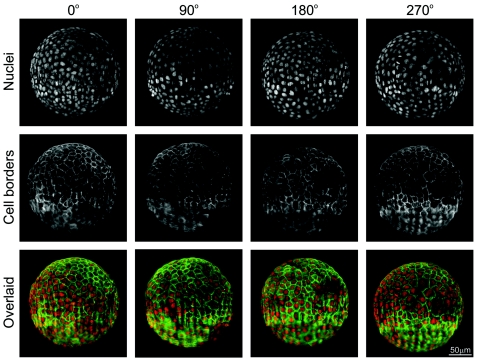
Light-sheet-based fluorescence microscopy (LSFM) is a fluorescence technique that combines optical sectioning, the key capability of confocal and two-photon fluorescence microscopes with multiple-view imaging, which is used in optical tomography. In contrast to conventional wide-field and confocal fluorescence microscopes, a light sheet illuminates only the focal plane of the detection objective lens from the side. Excitation is, thus, restricted to the fluorophores in the volume near the focal plane. This provides optical sectioning and allows the use of regular cameras in the detection process. Compared to confocal fluorescence microscopy, LSFM reduces photo bleaching and photo toxicity by up to three orders of magnitude. In LSFM, the specimen is embedded in a transparent block of hydrogel and positioned relative to the stationary light sheet using precise motorized translation and rotation stages. This feature is used to image any plane in a specimen. Additionally, multiple views obtained along different angles can be combined into a single data set with an improved resolution. LSFMs are very well suited for imaging large live specimens over long periods of time. However, they also perform well with very small specimens such as single yeast cells. This perspective introduces the principles of LSFM, explains the challenges of specimen preparation, and introduces the basics of a microscopy that takes advantage of multiple views.
Figures



References
-
- Colombelli, J, Pepperkok, R, Stelzer, E H, and Reynaud, E G, (2006). “La nanochirurgie laser.” Médecine Sciences, 22(6–7), 651–658. - PubMed
-
- Colombelli, J, Reynaud, E G, Rietdorf, J, Pepperkok, R, and Stelzer, E H (2005). “In vivo selective cytoskeleton dynamics quantification in interphase cells induced by pulsed ultraviolet laser nanosurgery.” Traffic 6, 1093–1102. - PubMed
-
- Dodt, H U, Leischner, U, Schierloh, A, Jährling, N, Mauch, C P, Deininger, K, Deussing, J M, Eder, M, Zieglgänsberger, W, and Becker, K (2007). “Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain.” Nat. Methods 4, 331–336. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources