

A structural model of latent evolutionary potentials underlying neutral networks in proteins
- PMID: 19404462
- PMCID: PMC2645552
- DOI: 10.2976/1.2739116/10.2976/1
A structural model of latent evolutionary potentials underlying neutral networks in proteins
Abstract
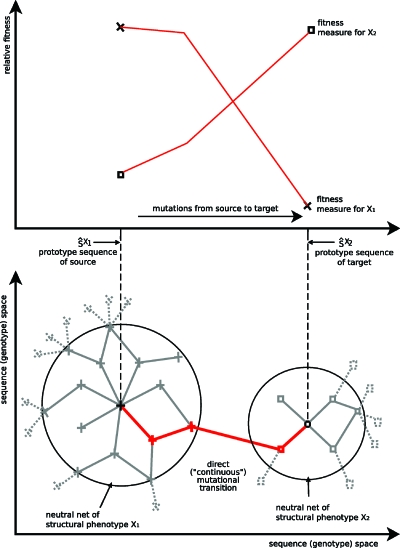
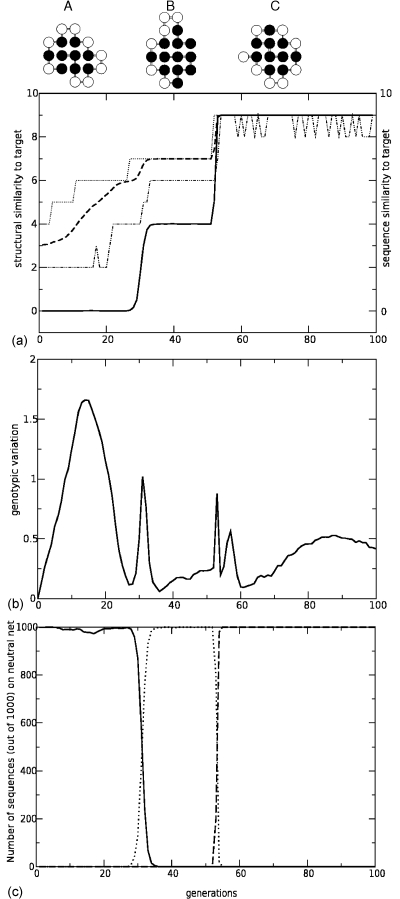
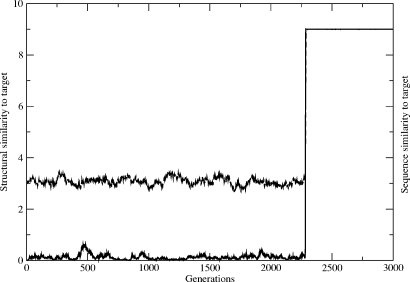
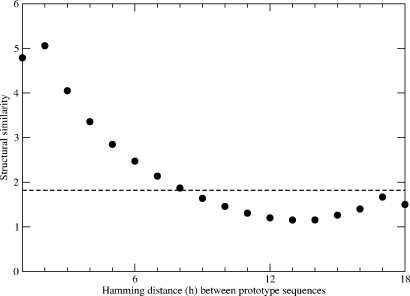
A central question in molecular evolution concerns the nature of phenotypic transitions, in particular, if neutral mutations hamper or somehow facilitate adaptability of proteins to new requirements. Proteins have been found to fluctuate between different structures, with frequencies of structures being proportional to their stability. Therefore, functional promiscuity may correspond to different structures with energies close to the ground state which then represent multiple selectable traits. We here postulate that these near-ground-state structures facilitate smooth transitions between phenotypes. Using a biophysical heteropolymer model with exhaustive mappings of sequences onto structures, we demonstrate that this is indeed possible because of a smooth gradient of stability along which any structural phenotype can be optimized and also because of mutational proximity of similar phenotypes in genotype space. Our model provides a biophysical rationalization of the intriguing, and otherwise puzzling experimental observation that adaptation to new requirements, e.g., latent function of a promiscuous enzyme, can proceed while the "old," phenotypically dominant function is maintained along a series of seemingly neutral mutations (see accompanying article). Thus pleiotropy may facilitate adaptation of latent traits before gene duplications and increase the effective adaptability of proteins.
Figures
References
-
- Aharoni, A, Gaidukov, L, Khersonsky, O, Gould, S M, Roodveldt, C, and Tawfik, DS (2005). “The ‘evolvability’ of promiscuous protein functions.” Nat. Genet. 37, 73–76. - PubMed
LinkOut - more resources
Full Text Sources