

Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease
- PMID: 19406126
- PMCID: PMC2764556
- DOI: 10.1016/j.yjmcc.2009.04.014
Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease
Abstract
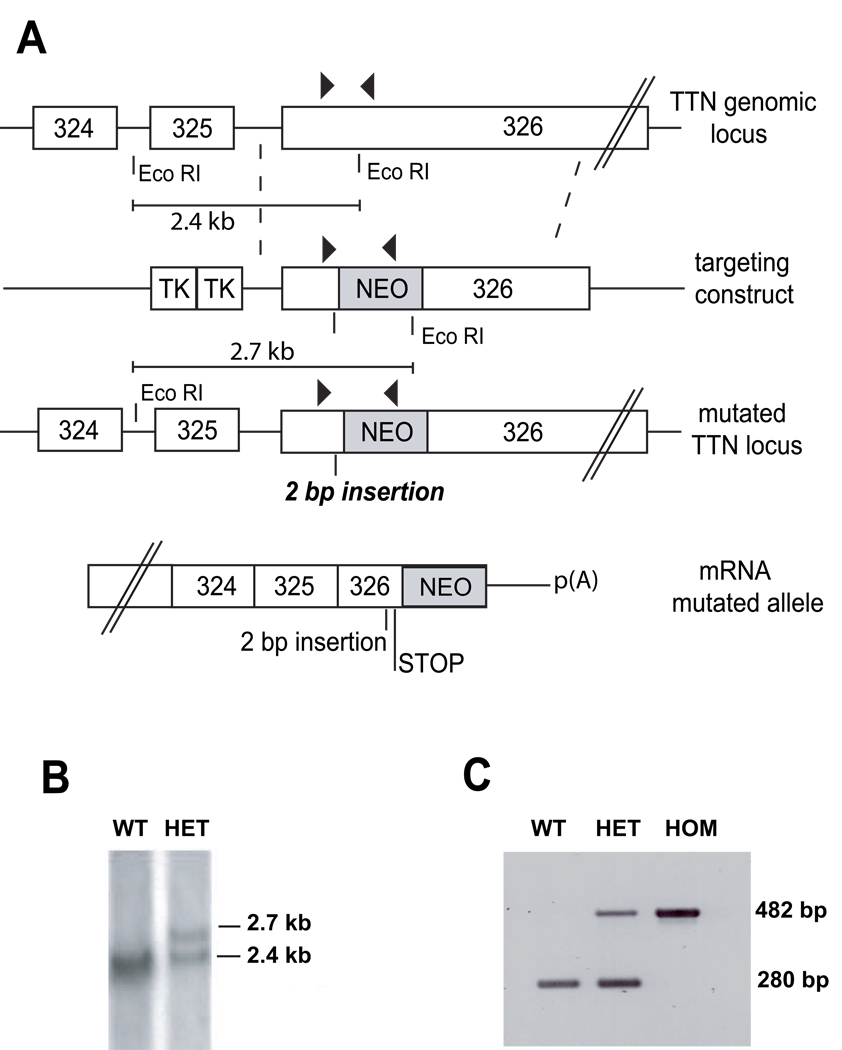
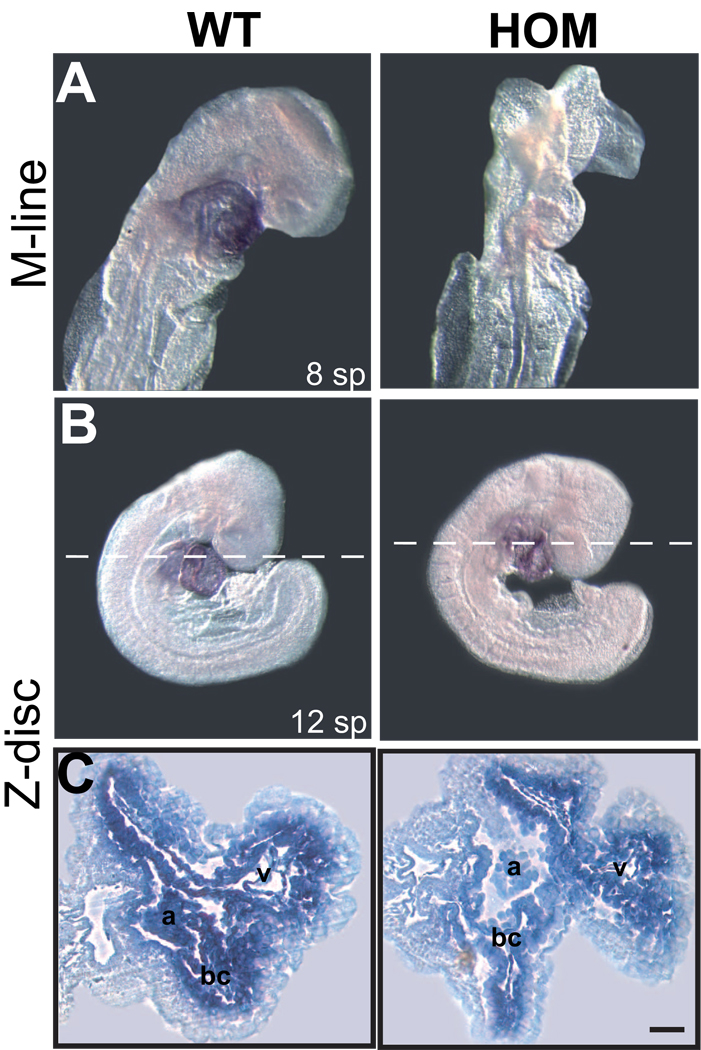
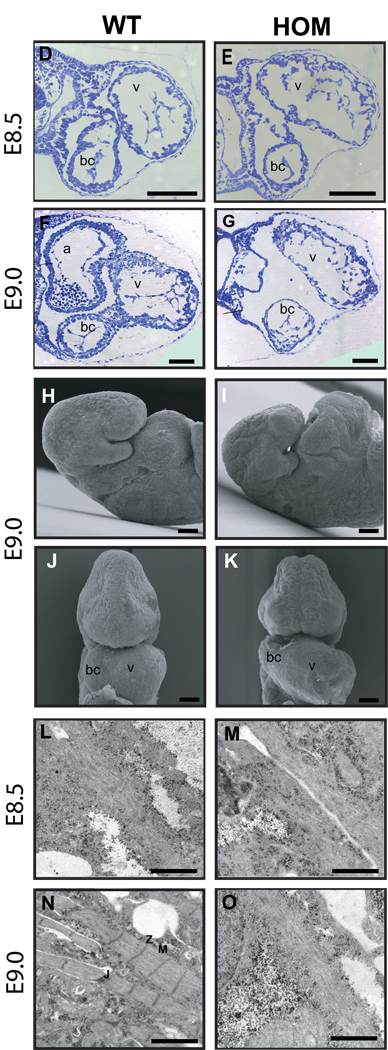
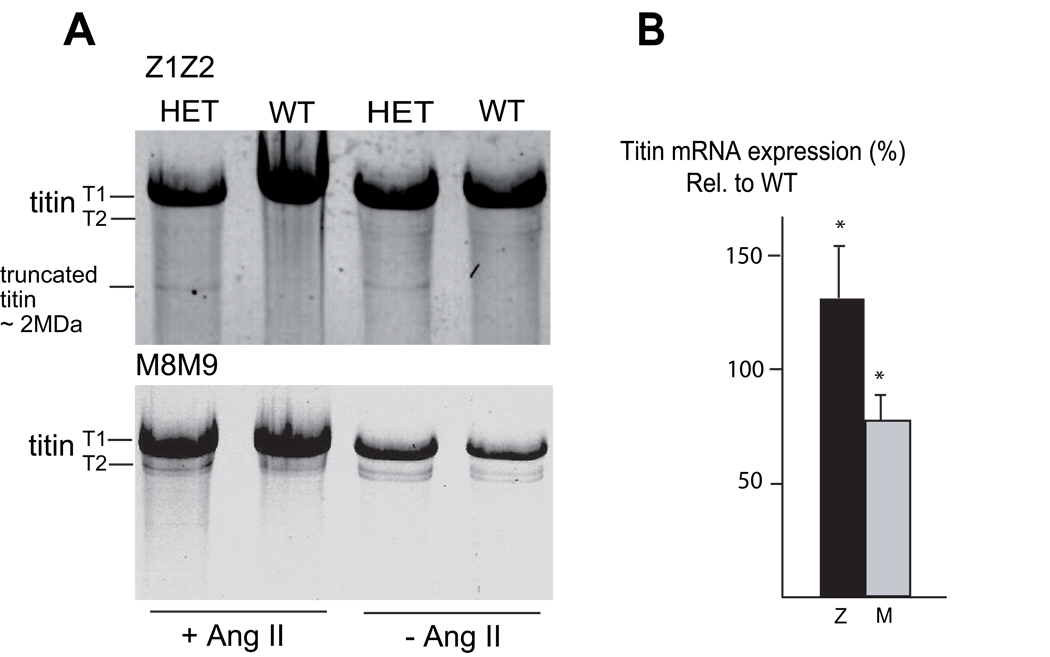
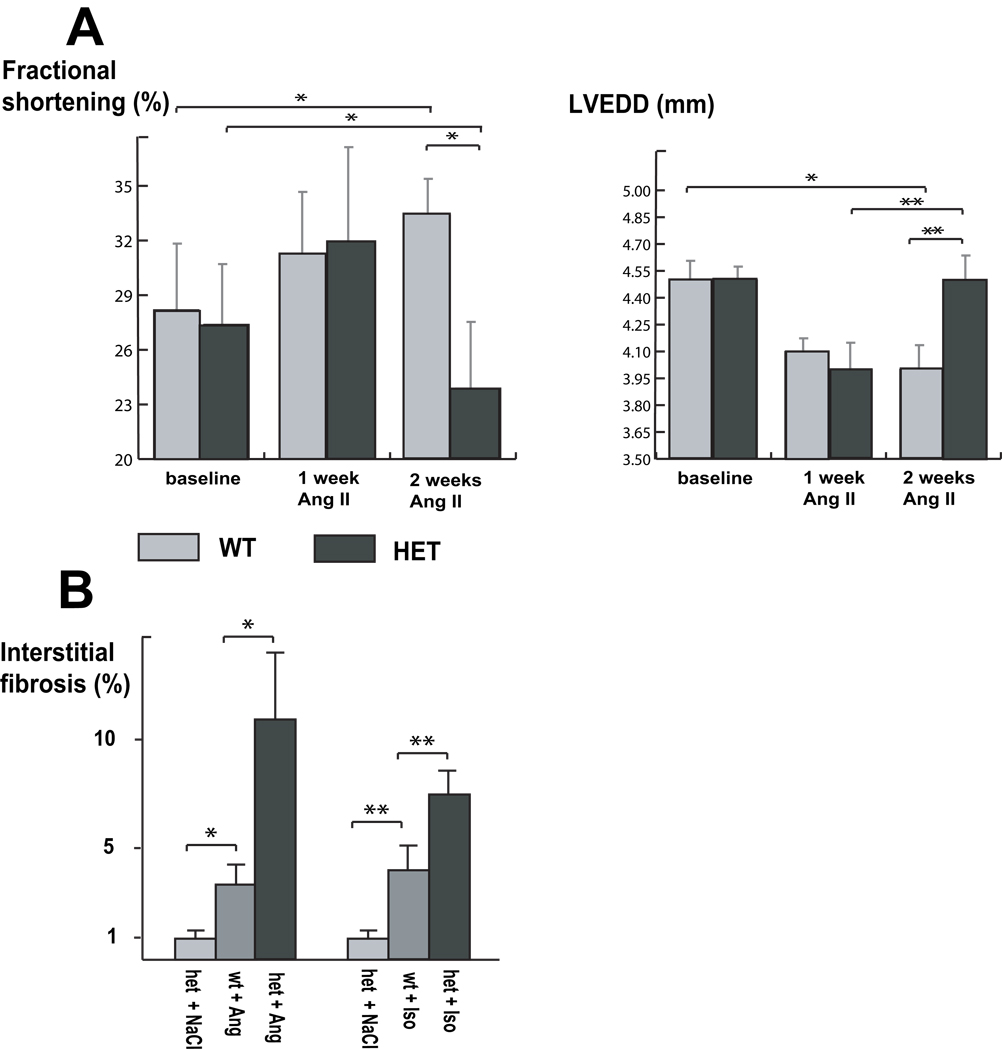
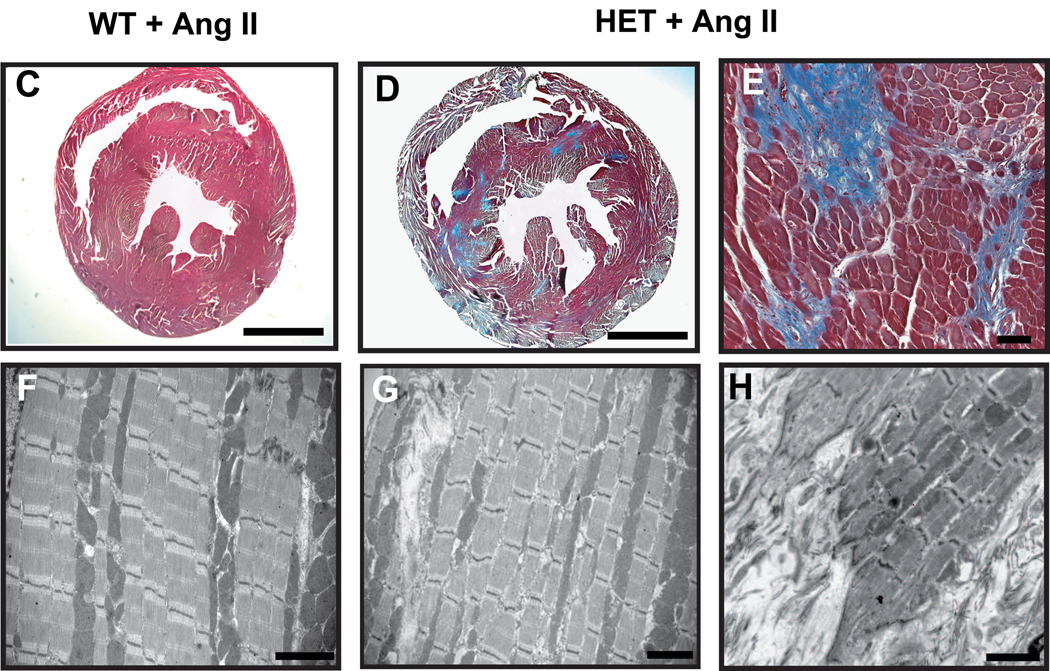
Mutations in a variety of myofibrillar genes cause dilated cardiomyopathy (DCM) in humans, usually with dominant inheritance and incomplete penetrance. Here, we sought to clarify the functional effects of the previously identified DCM-causing TTN 2-bp insertion mutation (c.43628insAT) and generated a titin knock-in mouse model mimicking the c.43628insAT allele. Mutant embryos homozygous for the Ttn knock-in mutation developed defects in sarcomere formation and consequently died before E9.5. Heterozygous mice were viable and demonstrated normal cardiac morphology, function and muscle mechanics. mRNA and protein expression studies on heterozygous hearts demonstrated elevated wild-type titin mRNA under resting conditions, suggesting that up-regulation of the wild-type titin allele compensates for the unstable mutated titin under these conditions. When chronically exposed to angiotensin II or isoproterenol, heterozygous mice developed marked left ventricular dilatation (p<0.05) with impaired fractional shortening (p<0.001) and diffuse myocardial fibrosis (11.95+/-2.8% vs. 3.7+/-1.1%). Thus, this model mimics typical features of human dilated cardiomyopathy and may further our understanding of how titin mutations perturb cardiac function and remodel the heart.
Conflict of interest statement
Figures
Comment in
-
Stressing the giant: a new approach to understanding dilated cardiomyopathy.J Mol Cell Cardiol. 2009 Sep;47(3):347-9. doi: 10.1016/j.yjmcc.2009.06.011. Epub 2009 Jun 22. J Mol Cell Cardiol. 2009. PMID: 19555694 Free PMC article. No abstract available.
References
-
- Dec GW, Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med. 1994;331:1564–1575. - PubMed
-
- Gerull B, Gramlich M, Atherton J, McNabb M, Trombitás K, Sasse-Klaassen S, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–204. - PubMed
-
- Gerull B, Atherton J, Geupel A, Sasse-Klaassen S, Heuser A, Frenneaux M, et al. Identification of a novel frameshift mutation in the giant muscle filament titin in a larger Australian family with dilated cardiomyopathy. J Mol M. 2006;84:478–483. - PubMed
-
- Satoh M, Takahashi M, Sakamoto T, Hiroe M, Marumo F, Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem Biophys Res Commun. 1999;262(2):411–417. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
