

The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency
- PMID: 19409522
- PMCID: PMC2681006
- DOI: 10.1016/j.ajhg.2009.04.004
The mitochondrial disulfide relay system protein GFER is mutated in autosomal-recessive myopathy with cataract and combined respiratory-chain deficiency
Abstract
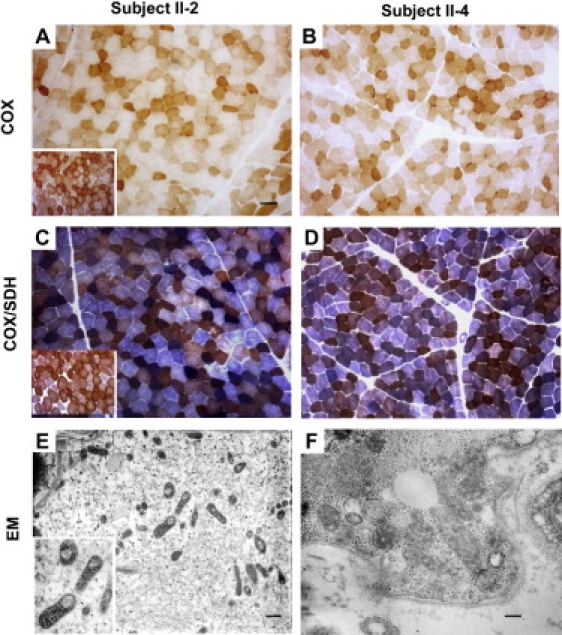
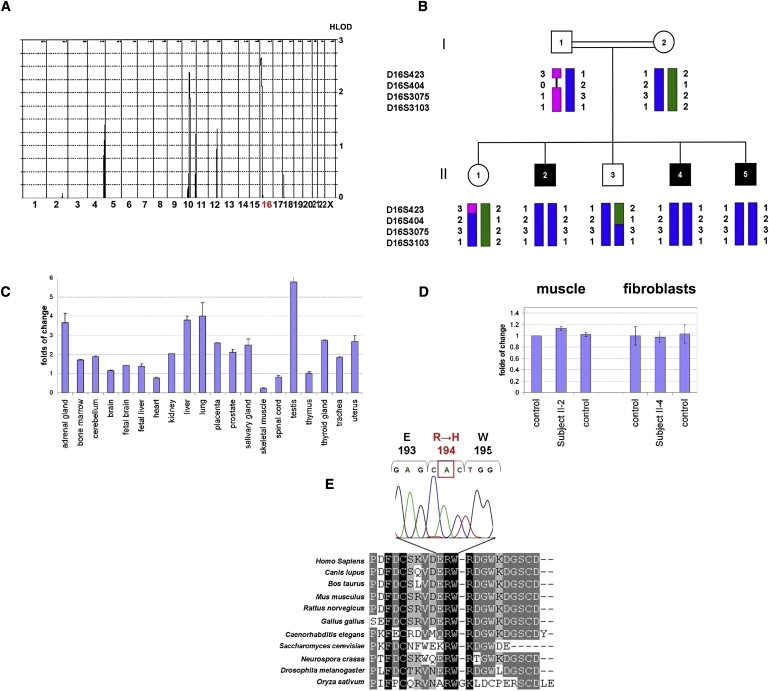
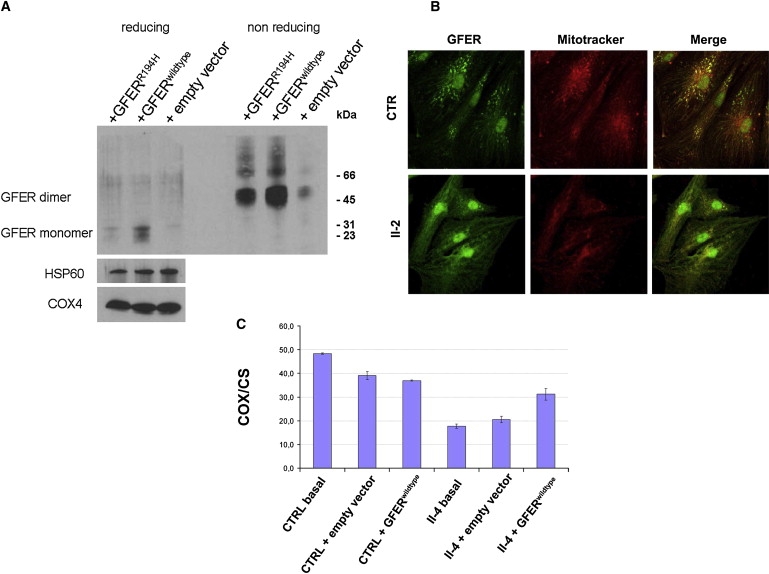
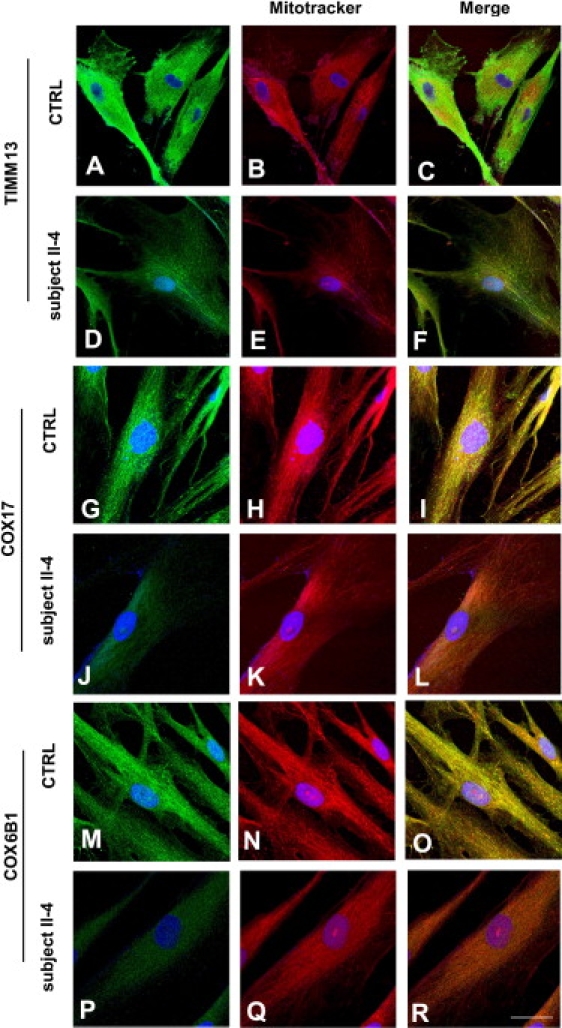
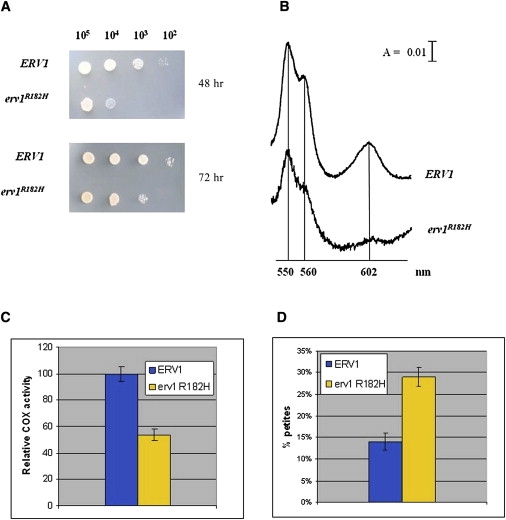
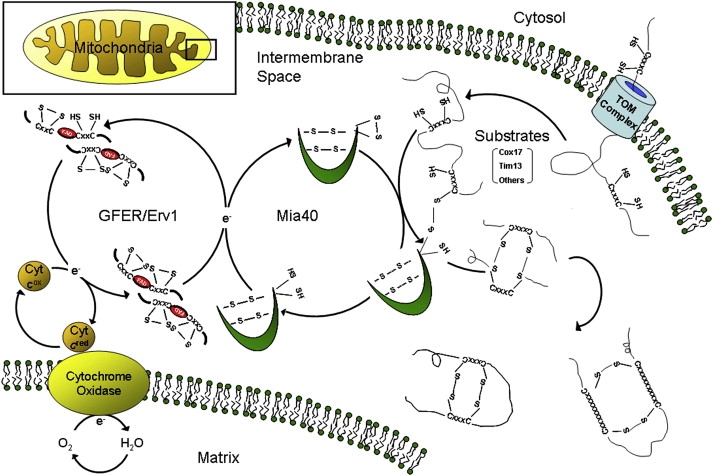
A disulfide relay system (DRS) was recently identified in the yeast mitochondrial intermembrane space (IMS) that consists of two essential components: the sulfhydryl oxidase Erv1 and the redox-regulated import receptor Mia40. The DRS drives the import of cysteine-rich proteins into the IMS via an oxidative folding mechanism. Erv1p is reoxidized within this system, transferring its electrons to molecular oxygen through interactions with cytochrome c and cytochrome c oxidase (COX), thereby linking the DRS to the respiratory chain. The role of the human Erv1 ortholog, GFER, in the DRS has been poorly explored. Using homozygosity mapping, we discovered that a mutation in the GFER gene causes an infantile mitochondrial disorder. Three children born to healthy consanguineous parents presented with progressive myopathy and partial combined respiratory-chain deficiency, congenital cataract, sensorineural hearing loss, and developmental delay. The consequences of the mutation at the level of the patient's muscle tissue and fibroblasts were 1) a reduction in complex I, II, and IV activity; 2) a lower cysteine-rich protein content; 3) abnormal ultrastructural morphology of the mitochondria, with enlargement of the IMS space; and 4) accelerated time-dependent accumulation of multiple mtDNA deletions. Moreover, the Saccharomyces cerevisiae erv1(R182H) mutant strain reproduced the complex IV activity defect and exhibited genetic instability of the mtDNA and mitochondrial morphological defects. These findings shed light on the mechanisms of mitochondrial biogenesis, establish the role of GFER in the human DRS, and promote an understanding of the pathogenesis of a new mitochondrial disease.
Figures
References
-
- DiMauro S., Schon E.A. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008;31:91–123. - PubMed
-
- Zeviani M., Carelli V. Mitochondrial disorders. Curr. Opin. Neurol. 2007;20:564–571. - PubMed
-
- Mesecke N., Terziyska N., Kozany C., Baumann F., Neupert W., Hell K., Herrmann J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. - PubMed
-
- Hell K. The Erv1-Mia40 disulfide relay system in the intermembrane space of mitochondria. Biochim. Biophys. Acta. 2008;1783:601–609. - PubMed
-
- Heckmatt J.Z., Dubowitz V. Needle biopsy of skeletal muscle. Muscle Nerve. 1984;7:594. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
