

Constructing genomic maps of positive selection in humans: where do we go from here?
- PMID: 19411596
- PMCID: PMC3647533
- DOI: 10.1101/gr.086652.108
Constructing genomic maps of positive selection in humans: where do we go from here?
Abstract
Identifying targets of positive selection in humans has, until recently, been frustratingly slow, relying on the analysis of individual candidate genes. Genomics, however, has provided the necessary resources to systematically interrogate the entire genome for signatures of natural selection. To date, 21 genome-wide scans for recent or ongoing positive selection have been performed in humans. A key challenge is to begin synthesizing these newly constructed maps of positive selection into a coherent narrative of human evolutionary history and derive a deeper mechanistic understanding of how natural populations evolve. Here, I chronicle the recent history of the burgeoning field of human population genomics, critically assess genome-wide scans for positive selection in humans, identify important gaps in knowledge, and discuss both short- and long-term strategies for traversing the path from the low-resolution, incomplete, and error-prone maps of selection today to the ultimate goal of a detailed molecular, mechanistic, phenotypic, and population genetics characterization of adaptive alleles.
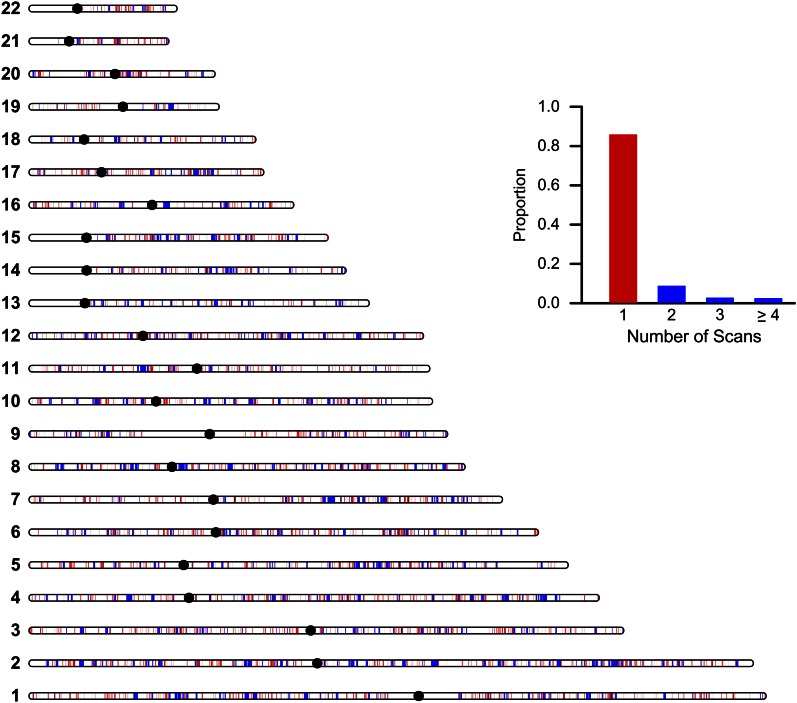
Figures


References
-
- Ahmed Z.M., Riazuddin S., Ahmad J., Bernstein S.L., Guo Y., Sabar M.F., Sieving P., Griffith A.J., Friedman T.B., Belyantseva I.A., et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 2003;12:3215–3223. - PubMed
-
- Akey J.M., Zhang K., Xiong M., Jin L. The effect of single nucleotide polymorphism identification strategies on estimates of linkage disequilibrium. Mol. Biol. Evol. 2003;20:232–242. - PubMed
-
- Akey J.M., Swanson W.J., Madeoy J., Eberle M., Shriver M.D. TRPV6 exhibits unusual patterns of polymorphism and divergence in worldwide populations. Hum. Mol. Genet. 2006;15:2106–2113. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources