

Prediction of protein binding regions in disordered proteins
- PMID: 19412530
- PMCID: PMC2671142
- DOI: 10.1371/journal.pcbi.1000376
Prediction of protein binding regions in disordered proteins
Abstract
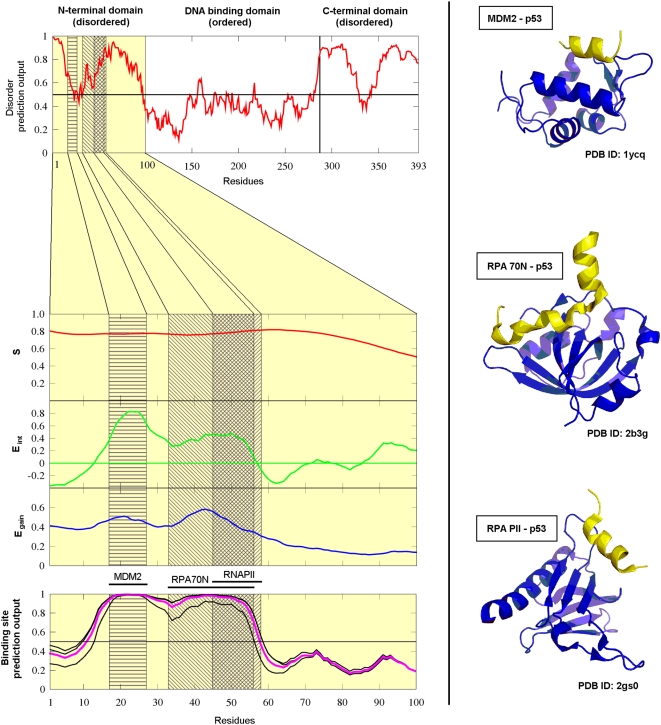
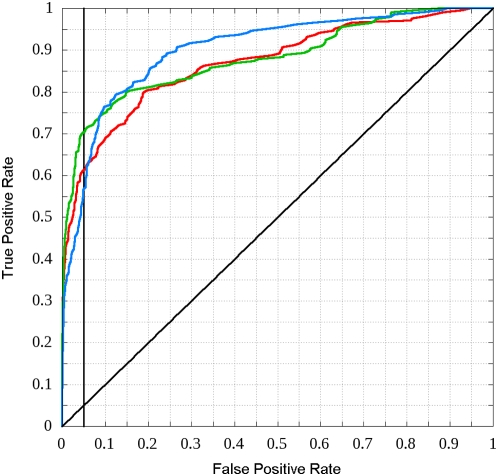
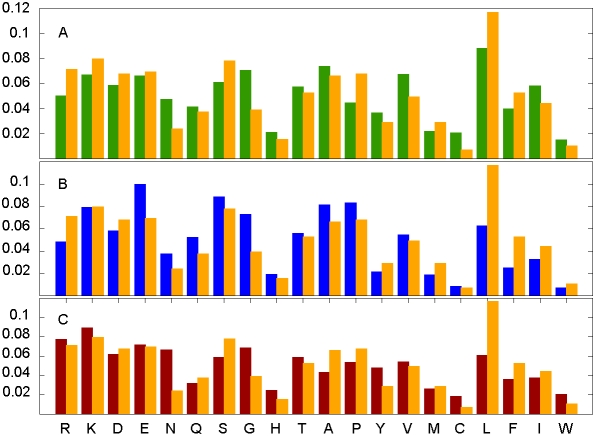
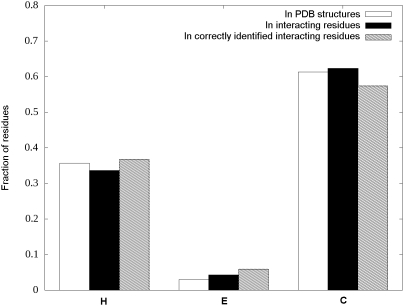
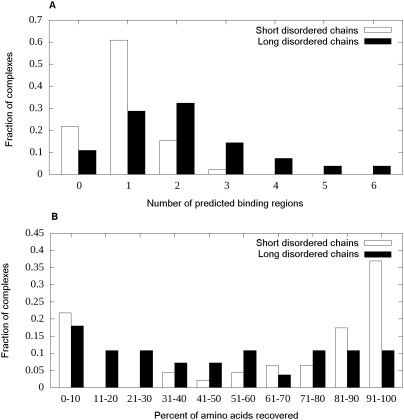
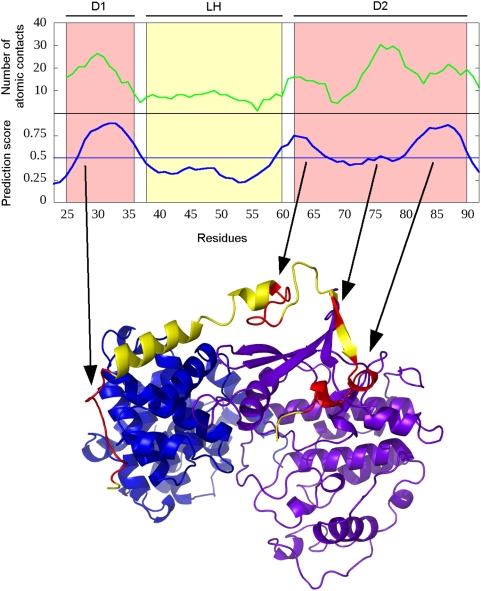
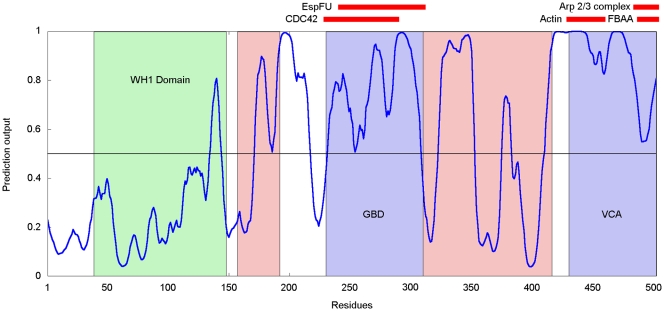
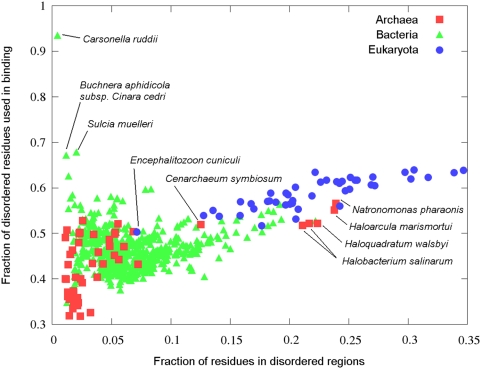
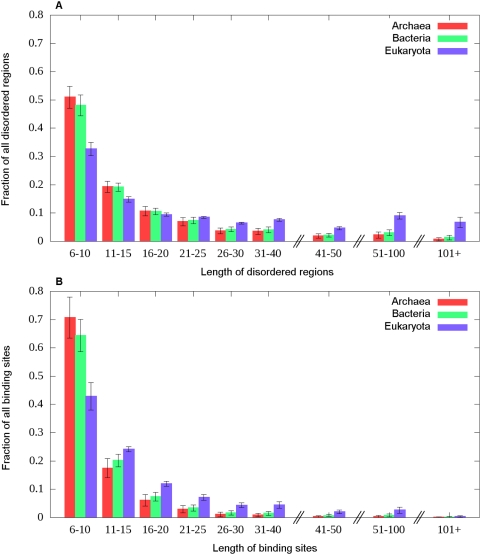
Many disordered proteins function via binding to a structured partner and undergo a disorder-to-order transition. The coupled folding and binding can confer several functional advantages such as the precise control of binding specificity without increased affinity. Additionally, the inherent flexibility allows the binding site to adopt various conformations and to bind to multiple partners. These features explain the prevalence of such binding elements in signaling and regulatory processes. In this work, we report ANCHOR, a method for the prediction of disordered binding regions. ANCHOR relies on the pairwise energy estimation approach that is the basis of IUPred, a previous general disorder prediction method. In order to predict disordered binding regions, we seek to identify segments that are in disordered regions, cannot form enough favorable intrachain interactions to fold on their own, and are likely to gain stabilizing energy by interacting with a globular protein partner. The performance of ANCHOR was found to be largely independent from the amino acid composition and adopted secondary structure. Longer binding sites generally were predicted to be segmented, in agreement with available experimentally characterized examples. Scanning several hundred proteomes showed that the occurrence of disordered binding sites increased with the complexity of the organisms even compared to disordered regions in general. Furthermore, the length distribution of binding sites was different from disordered protein regions in general and was dominated by shorter segments. These results underline the importance of disordered proteins and protein segments in establishing new binding regions. Due to their specific biophysical properties, disordered binding sites generally carry a robust sequence signal, and this signal is efficiently captured by our method. Through its generality, ANCHOR opens new ways to study the essential functional sites of disordered proteins.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. - PubMed
-
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. - PubMed
-
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, et al. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. - PubMed
-
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. - PubMed
-
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
