

Willows: a memory efficient tree and forest construction package
- PMID: 19416535
- PMCID: PMC2683818
- DOI: 10.1186/1471-2105-10-130
Willows: a memory efficient tree and forest construction package
Abstract
Background: Existing tree and forest methods are powerful bioinformatics tools to explore high dimensional data including high throughput genomic data. However, they cannot deal with the data generated by recent genotyping platforms for single nucleotide polymorphisms due to the massive size of the data and its excessive memory demand.
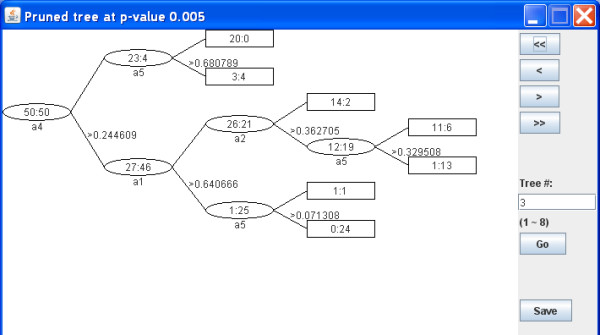
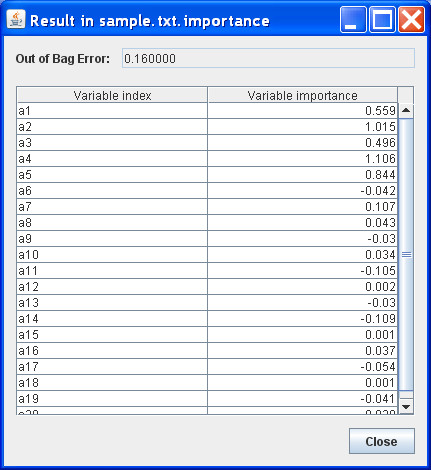
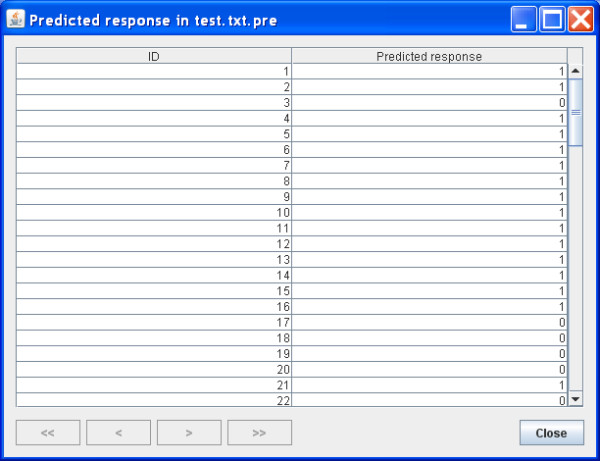
Results: Using the recursive partitioning technique, we developed a new software package, Willows, to maximize the utility of the computer memory and make it feasible to analyze massive genotype data. This package includes three tree-based methods -- classification tree, random forest, and deterministic forest, and can efficiently handle the massive amount of SNP data. In addition, this package can easily set different options (e.g., algorithms and specifications) and predict the class of test samples.
Conclusion: We developed Willows in a user friendly interface with the goal of maximizing the use of memory, which is critical for analysis of genomic data. The Willows package is well documented and publicly available at (http://c2s2.yale.edu/software/Willows).
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
