

Elucidation of separate, but collaborative functions of the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 in mitochondrial biogenesis reveals new insight into maternally inherited deafness
- PMID: 19417006
- PMCID: PMC2701340
- DOI: 10.1093/hmg/ddp208
Elucidation of separate, but collaborative functions of the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 in mitochondrial biogenesis reveals new insight into maternally inherited deafness
Abstract
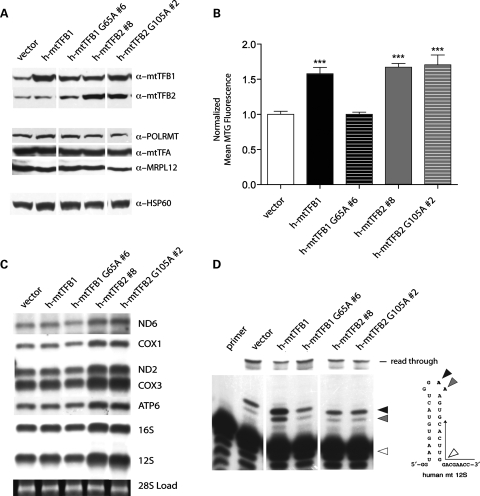
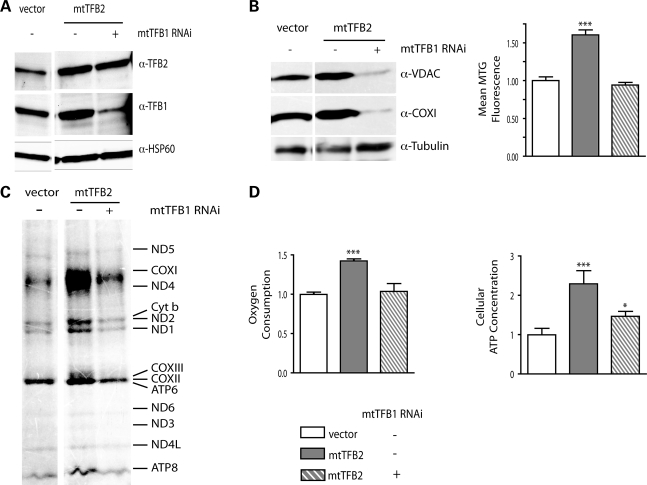
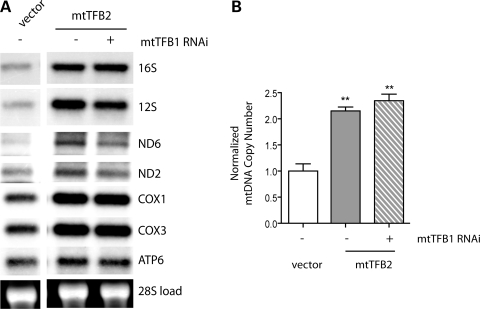
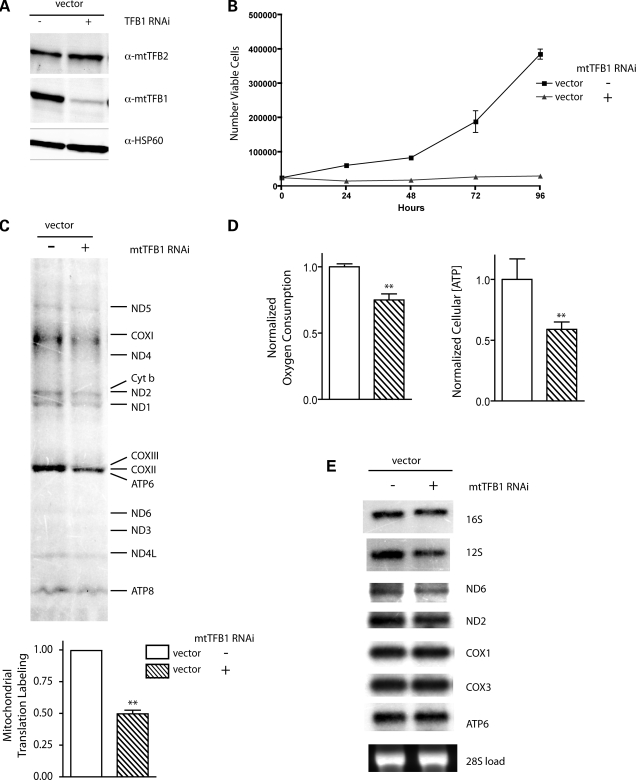
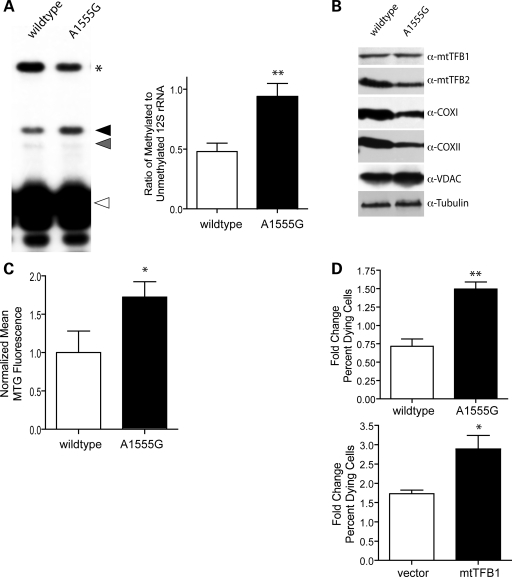
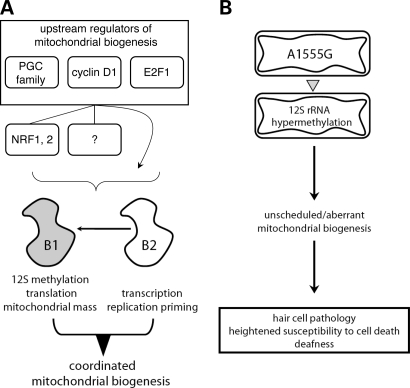
Mitochondrial biogenesis is controlled by signaling networks that relay information to and from the organelles. However, key mitochondrial factors that mediate such pathways and how they contribute to human disease are not understood fully. Here we demonstrate that the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 are key downstream effectors of mitochondrial biogenesis that perform unique, yet cooperative functions. The primary function of h-mtTFB2 is mtDNA transcription and maintenance, which is independent of its rRNA methyltransferase activity, while that of h-mtTFB1 is mitochondrial 12S rRNA methylation needed for normal mitochondrial translation, metabolism and cell growth. Over-expression of h-mtTFB1 causes 12S rRNA hypermethylation, aberrant mitochondrial biogenesis and increased sorbitol-induced cell death. These phenotypes are recapitulated in cells harboring the pathogenic A1555G mtDNA mutation, implicating a deleterious rRNA methylation-dependent retrograde signal in maternally inherited deafness pathology and shedding significant insight into how h-mtTFB1 acts as a nuclear modifier of this disease.
Figures
References
-
- Bonawitz N.D., Clayton D.A., Shadel G.S. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol. Cell. 2006;24:813–825. - PubMed
-
- Butow R.A., Avadhani N.G. Mitochondrial signaling: the retrograde response. Mol. Cell. 2004;14:1–15. - PubMed
-
- Scarpulla R.C. Transcriptional paradigms in Mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008;88:611–638. - PubMed
-
- Handschin C., Spiegelman B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr. Rev. 2006;27:728–735. - PubMed
-
- Asin-Cayuela J., Gustafsson C.M. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci. 2007;32:111–117. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
