

Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis
- PMID: 19417144
- PMCID: PMC2889001
- DOI: 10.1158/1535-7163.MCT-08-1166
Transcriptional induction of GRP78/BiP by histone deacetylase inhibitors and resistance to histone deacetylase inhibitor-induced apoptosis
Abstract
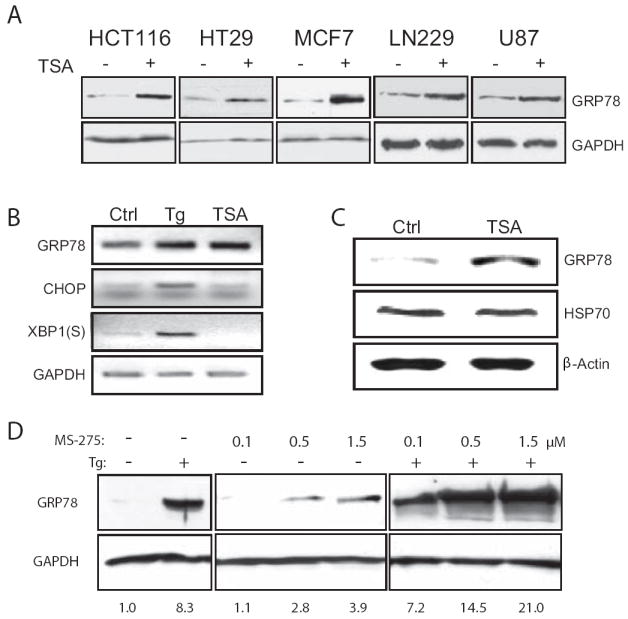
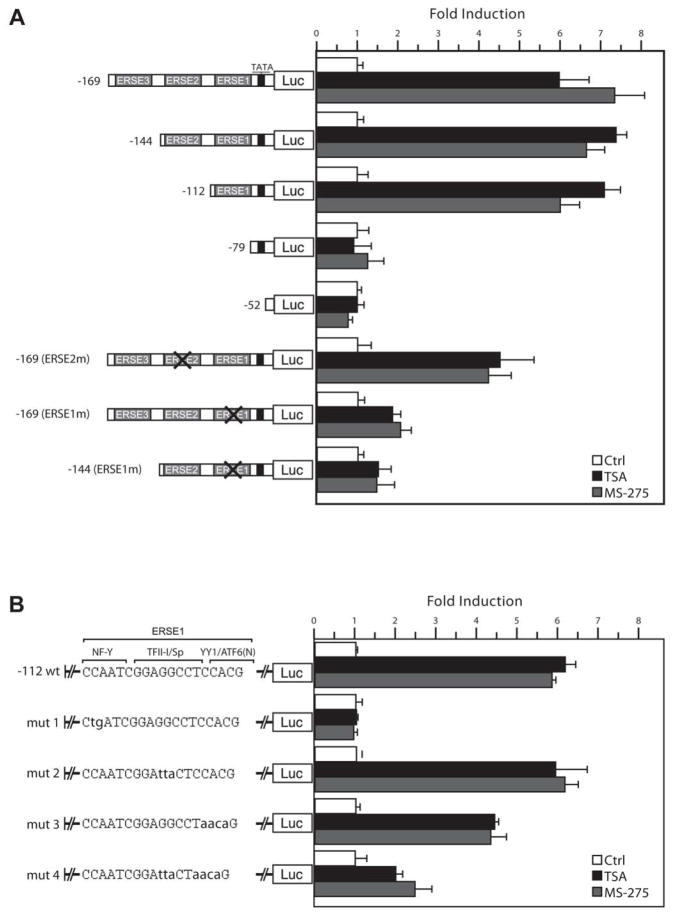
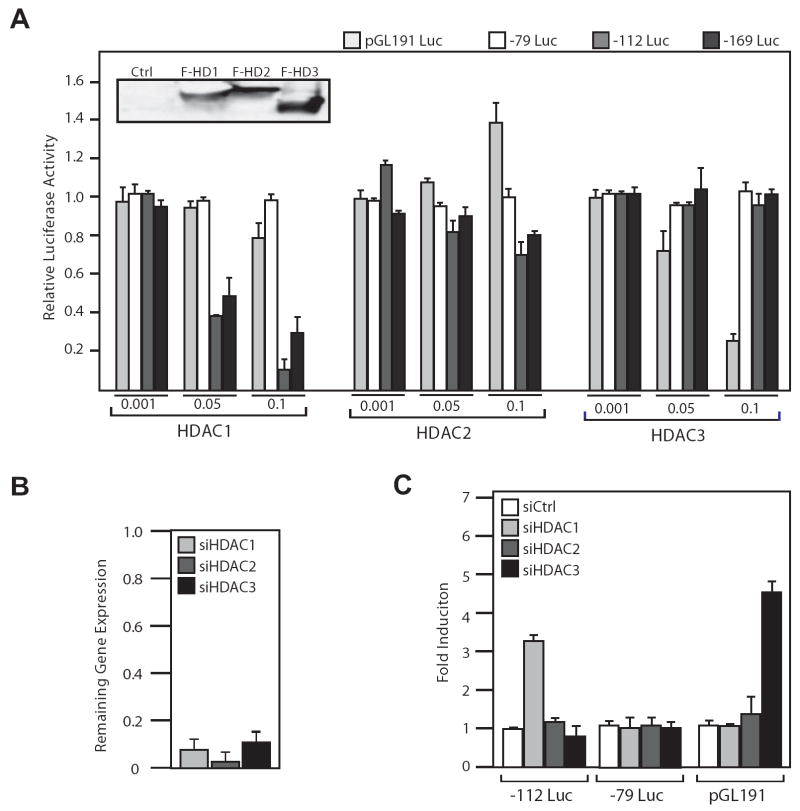
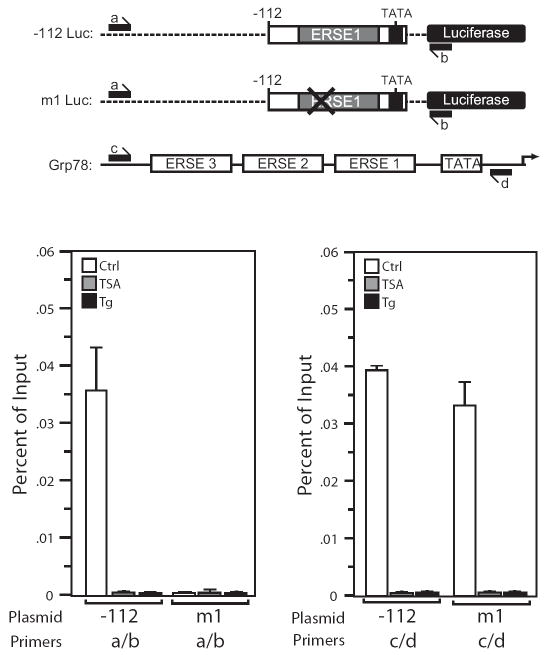
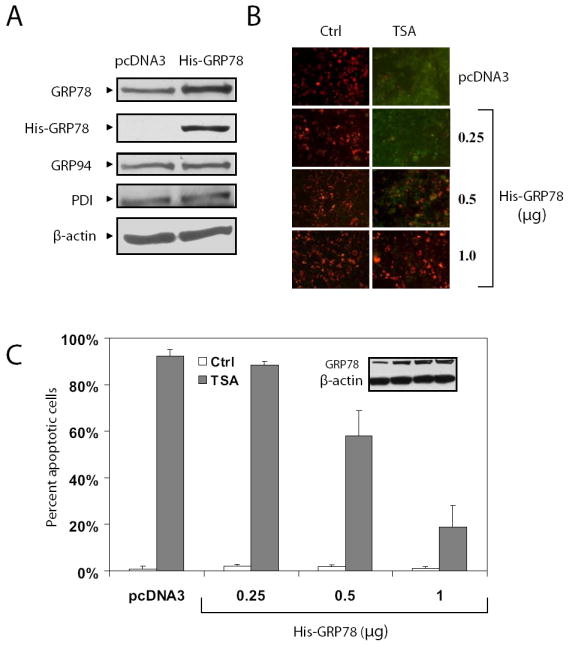
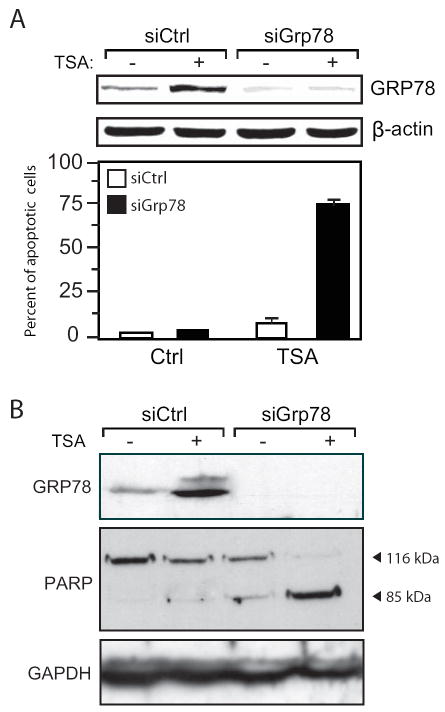
Histone deacetylase (HDAC) inhibitors are emerging as effective therapies in the treatment of cancer, and the role of HDACs in the regulation of promoters is rapidly expanding. GRP78/BiP is a stress inducible endoplasmic reticulum (ER) chaperone with antiapoptotic properties. We present here the mechanism for repression of the Grp78 promoter by HDAC1. Our studies reveal that HDAC inhibitors specifically induce GRP78, and the induction level is amplified by ER stress. Through mutational analysis, we have identified the minimal Grp78 promoter and specific elements responsible for HDAC-mediated repression. We show the involvement of HDAC1 in the negative regulation of the Grp78 promoter not only by its induction in the presence of the HDAC inhibitors trichostatin A and MS-275 but also by exogenous overexpression and small interfering RNA knockdown of specific HDACs. We present the results of chromatin immunoprecipitation analysis that reveals the binding of HDAC1 to the Grp78 promoter before, but not after, ER stress. Furthermore, overexpression of GRP78 confers resistance to HDAC inhibitor-induced apoptosis in cancer cells, and conversely, suppression of GRP78 sensitizes them to HDAC inhibitors. These results define HDAC inhibitors as new agents that up-regulate GRP78 without concomitantly inducing the ER or heat shock stress response, and suppression of GRP78 in tumors may provide a novel, adjunctive option to enhance anticancer therapies that use these compounds.
Figures
References
-
- Glozak M, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. - PubMed
-
- Yang X, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–8. - PubMed
-
- Wang J, Bown C, Young L. Differential display PCR reveals novel targets for the mood-stabilizing drug valproate including the molecular chaperone GRP78. Mol Pharmacol. 1999;55:521–7. - PubMed
-
- Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–10. - PubMed
-
- Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–97. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
