

Valproate activates the Notch3/c-FLIP signaling cascade: a strategy to attenuate white matter hyperintensities in bipolar disorder in late life?
- PMID: 19419383
- PMCID: PMC2788821
- DOI: 10.1111/j.1399-5618.2009.00675.x
Valproate activates the Notch3/c-FLIP signaling cascade: a strategy to attenuate white matter hyperintensities in bipolar disorder in late life?
Abstract
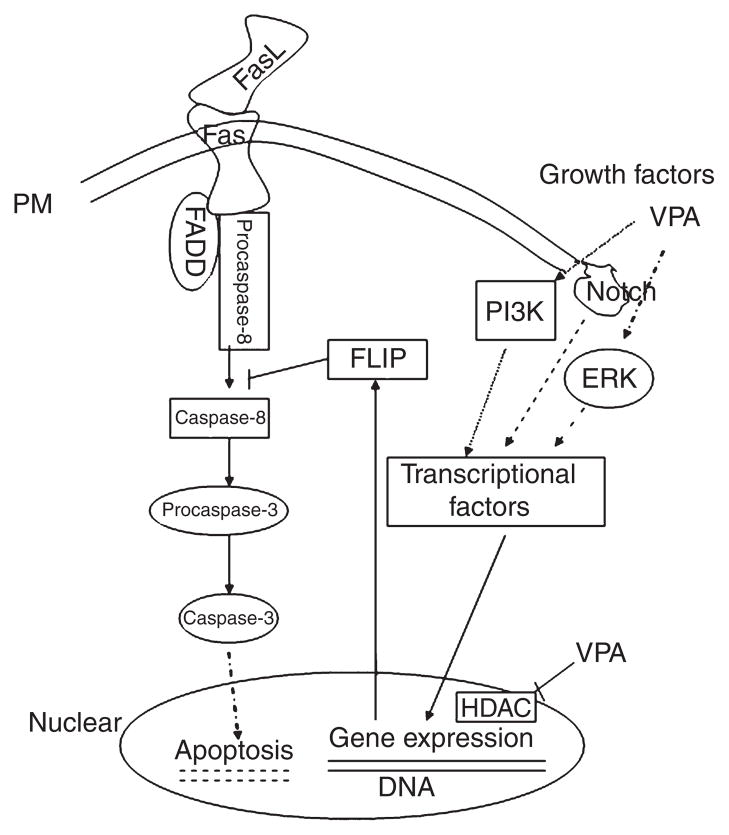
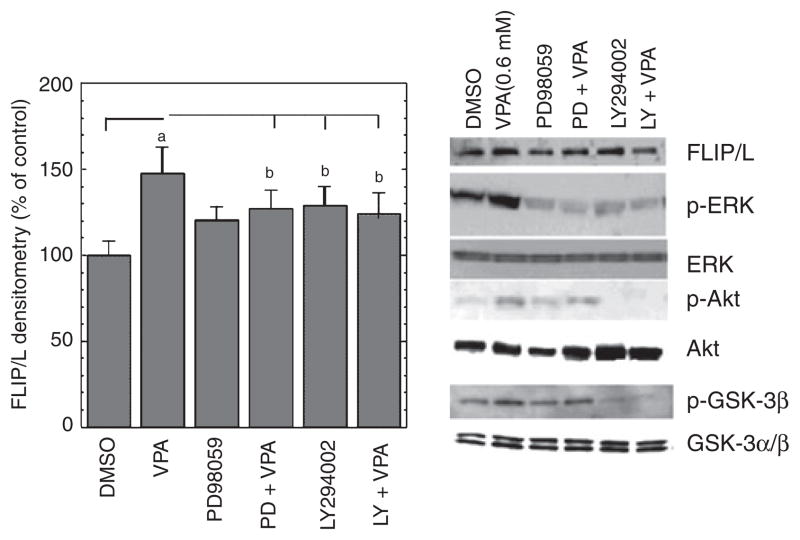
Objectives: Increased prevalence of deep white matter hyperintensities (DWMHs) has been consistently observed in patients with geriatric depression and bipolar disorder. DMWHs are associated with chronicity, disability, and poor quality of life. They are thought to be ischemic in their etiology and may be related to the underlying pathophysiology of mood disorders in the elderly. Notably, these lesions strikingly resemble radiological findings related to the cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephelopathy (CADASIL) syndrome. CADASIL arises from mutations in Notch3, resulting in impaired signaling via cellular Fas-associated death domain-like interleukin-1-beta-converting enzyme-inhibitory protein (c-FLIP) through an extracellular signal-regulated kinase (ERK)-dependent pathway. These signaling abnormalities have been postulated to underlie the progressive degeneration of vascular smooth muscle cells (VSMC). This study investigates the possibility that the anticonvulsant valproate (VPA), which robustly activates the ERK mitogen-activated protein kinase (MAPK) cascade, may exert cytoprotective effects on VSMC through the Notch3/c-FLIP pathway.
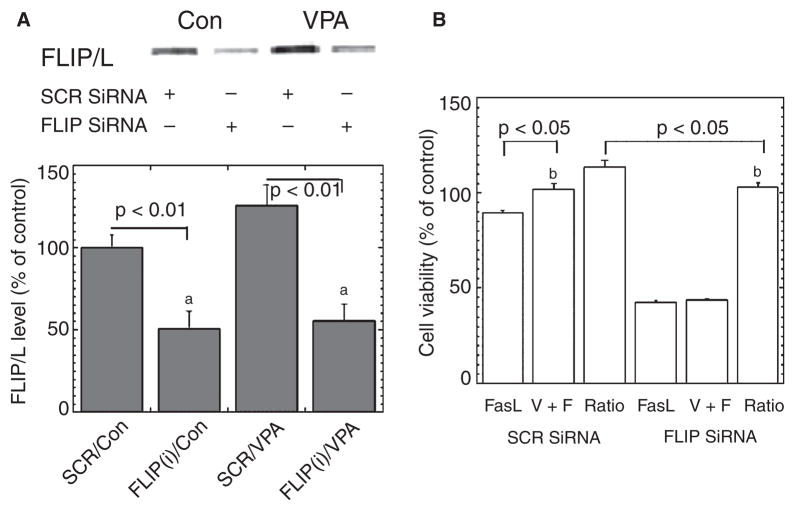
Methods: Human VSMC were treated with therapeutic concentrations of VPA subchronically. c-FLIP was knocked down via small interfering ribonucleic acid transfection. Cell survival, apoptosis, and protein levels were measured.
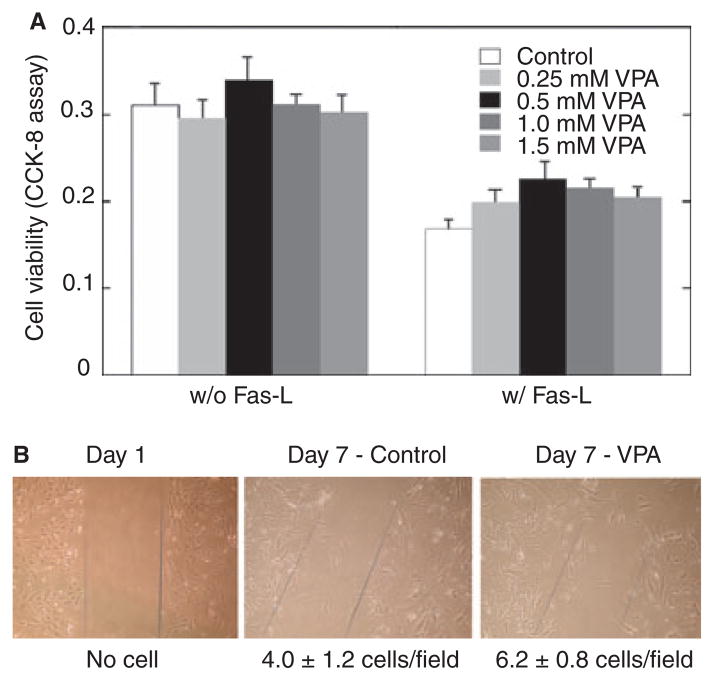
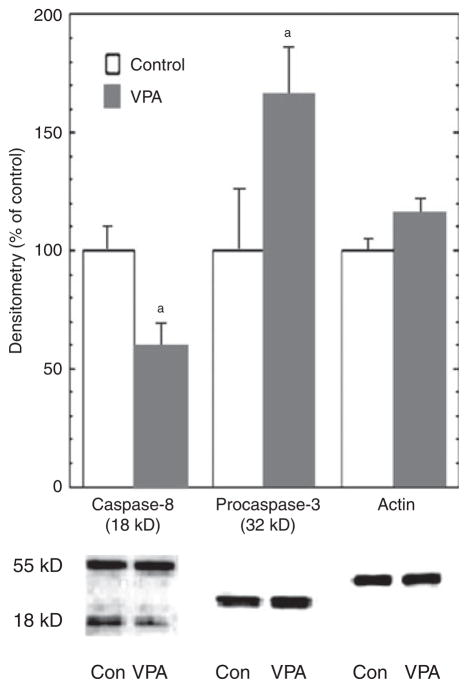
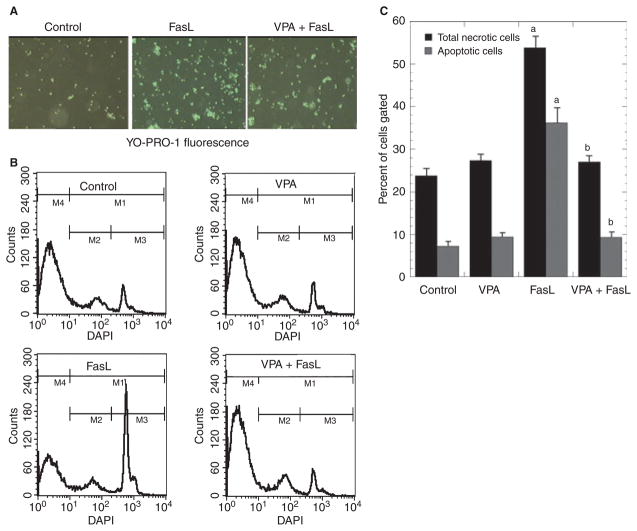
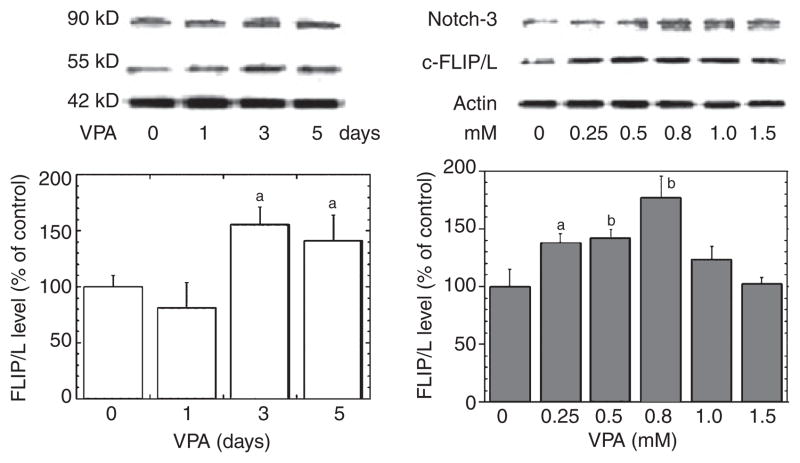
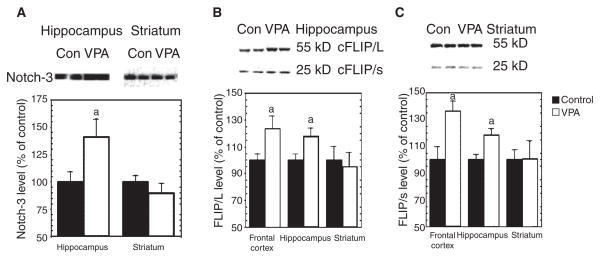
Results: VPA increased c-FLIP levels dose- and time-dependently and promoted VSMC survival in response to Fas ligand-induced apoptosis in VSMC. The anti-apoptotic effect of VPA was abolished by c-FLIP knockdown. VPA also produced similar in vivo effects in rat brain.
Conclusions: These results raise the intriguing possibility that VPA may be a novel therapeutic agent for the treatment of CADASIL and related disorders. They also suggest that VPA might decrease the liability of patients with late-life mood disorders to develop DWMHs.
Figures
References
-
- Videbech P. MRI findings in patients with affective disorder: a meta-analysis. Acta Psychiatr Scand. 1997;96:157–168. - PubMed
-
- Altshuler LL, Curran JG, Hauser P, Mintz J, Denico K, Post R. T-2 hyperintensities in bipolar disorder – magnetic-resonance- imaging comparison and literature metaanalysis. Am J Psychiatry. 1995;152:1139–1144. - PubMed
-
- Thomas AJ, Perry R, Kalaria RN, Oakley A, McMeekin W, O’Brien JT. Pathologies and pathological mechanisms for white matter hyperintensities in depression. Ann N Y Acad Sci. 2002;977:333–339. - PubMed
-
- Thomas AJ, O’Brien JT, Davis S, et al. Ischemic basis for deep white matter hyperintensities in major depression – a neuropathological study. Arch Gen Psychiatry. 2002;59:785–792. - PubMed
-
- Ruchoux MM, Guerouaou D, Vandenhaute B, Pruvo JP, Vermersch P, Leys D. Systemic vascular smooth muscle cell impairment in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Acta Neuropathol (Berl) 1995;89:500–512. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
