

Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage
- PMID: 19420089
- PMCID: PMC2702835
- DOI: 10.1093/brain/awp102
Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage
Abstract
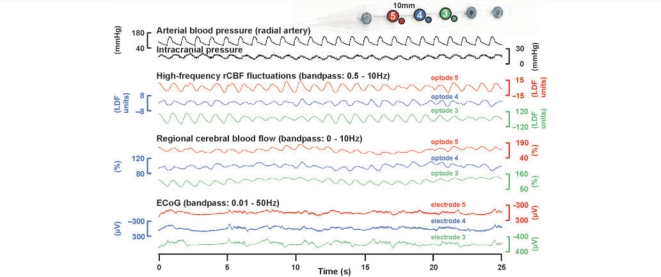
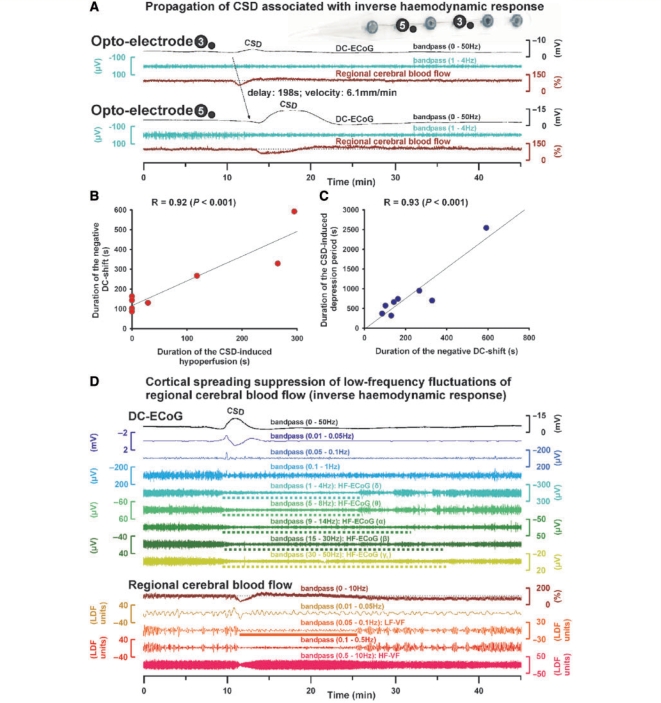
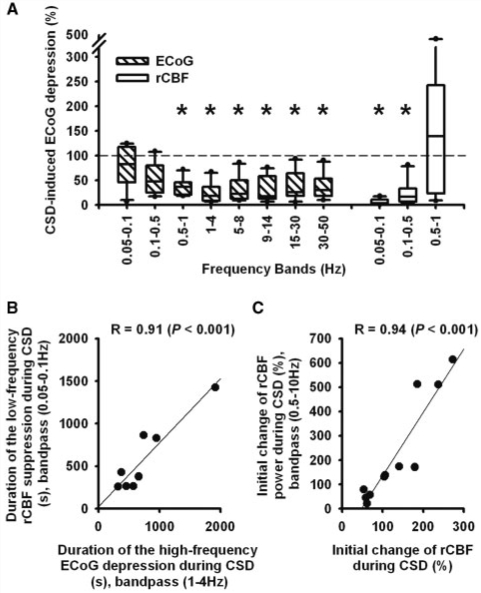
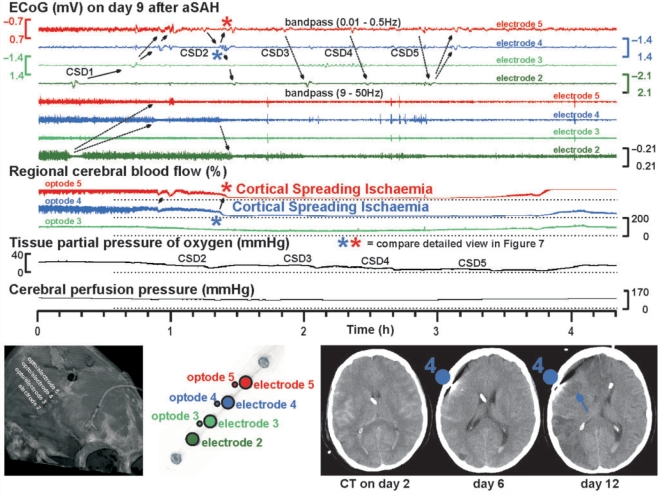
The term cortical spreading depolarization (CSD) describes a wave of mass neuronal depolarization associated with net influx of cations and water. Clusters of prolonged CSDs were measured time-locked to progressive ischaemic damage in human cortex. CSD induces tone alterations in resistance vessels, causing either transient hyperperfusion (physiological haemodynamic response) in healthy tissue; or hypoperfusion [inverse haemodynamic response = cortical spreading ischaemia (CSI)] in tissue at risk for progressive damage, which has so far only been shown experimentally. Here, we performed a prospective, multicentre study in 13 patients with aneurysmal subarachnoid haemorrhage, using novel subdural opto-electrode technology for simultaneous laser-Doppler flowmetry (LDF) and direct current-electrocorticography, combined with measurements of tissue partial pressure of oxygen (ptiO(2)). Regional cerebral blood flow and electrocorticography were simultaneously recorded in 417 CSDs. Isolated CSDs occurred in 12 patients and were associated with either physiological, absent or inverse haemodynamic responses. Whereas the physiological haemodynamic response caused tissue hyperoxia, the inverse response led to tissue hypoxia. Clusters of prolonged CSDs were measured in five patients in close proximity to structural brain damage as assessed by neuroimaging. Clusters were associated with CSD-induced spreading hypoperfusions, which were significantly longer in duration (up to 144 min) than those of isolated CSDs. Thus, oxygen depletion caused by the inverse haemodynamic response may contribute to the establishment of clusters of prolonged CSDs and lesion progression. Combined electrocorticography and perfusion monitoring also revealed a characteristic vascular signature that might be used for non-invasive detection of CSD. Low-frequency vascular fluctuations (LF-VF) (f < 0.1 Hz), detectable by functional imaging methods, are determined by the brain's resting neuronal activity. CSD provides a depolarization block of the resting activity, recorded electrophysiologically as spreading depression of high-frequency-electrocorticography activity. Accordingly, we observed a spreading suppression of LF-VF, which accompanied spreading depression of high-frequency-electrocorticography activity, independently of whether CSD was associated with a physiological, absent or inverse haemodynamic response. Spreading suppressions of LF-VF thus allow the differentiation of progressive ischaemia and repair phases in a fashion similar to that shown previously for spreading depressions of high-frequency-electrocorticography activity. In conclusion, it is suggested that (i) CSI is a novel human disease mechanism associated with lesion development and a potential target for therapeutic intervention in stroke; and that (ii) prolonged spreading suppressions of LF-VF are a novel 'functional marker' for progressive ischaemia.
Figures



References
-
- Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25:621–5. - PubMed
-
- Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and KCl-induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab. 1994;14:12–9. - PubMed
-
- Barth M, Capelle HH, Weidauer S, Weiss C, Münch E, Thomé C, et al. Effect of nicardipine prolonged-release implants on cerebral vasospasm and clinical outcome after severe aneurysmal subarachnoid hemorrhage: a prospective, randomized, double-blind phase IIa study. Stroke. 2007;38:330–6. - PubMed
-
- Biswal B, Yetkin FZ, Haughton VM, Hyde JS. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med. 1995;34:537–41. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
