

Widespread genomic signatures of natural selection in hominid evolution
- PMID: 19424416
- PMCID: PMC2669884
- DOI: 10.1371/journal.pgen.1000471
Widespread genomic signatures of natural selection in hominid evolution
Abstract
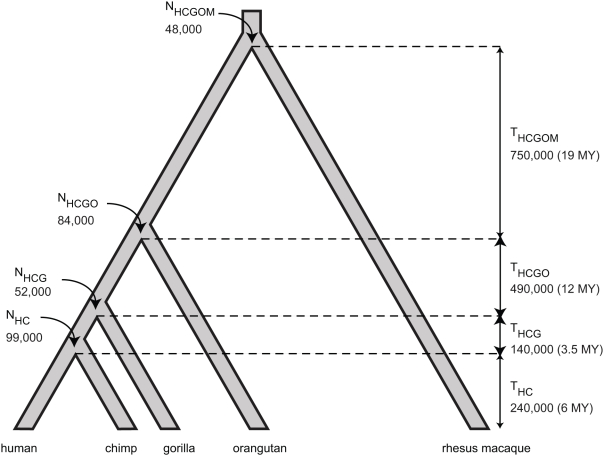
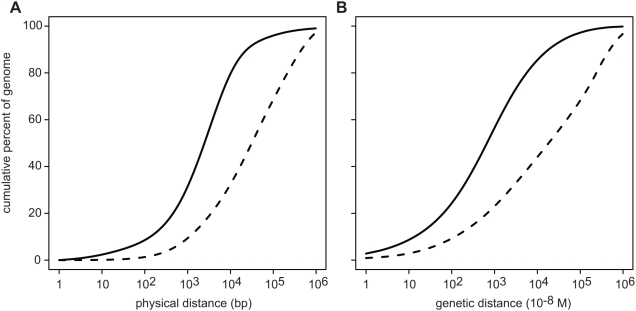
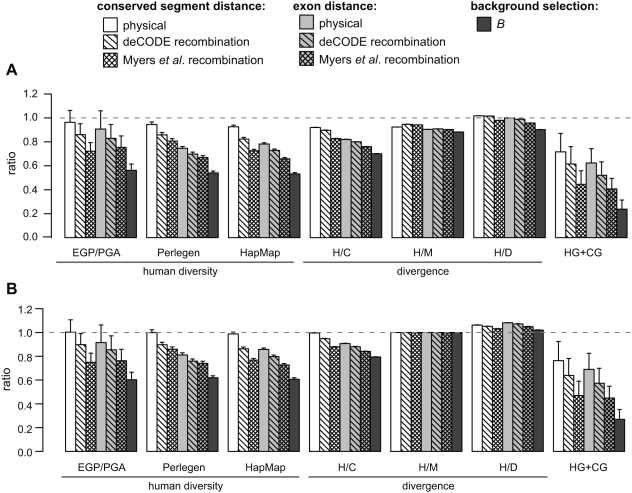
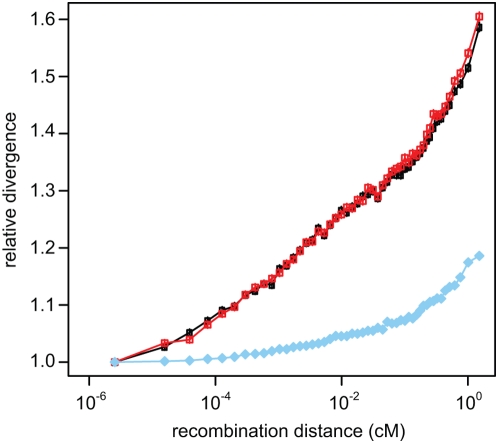
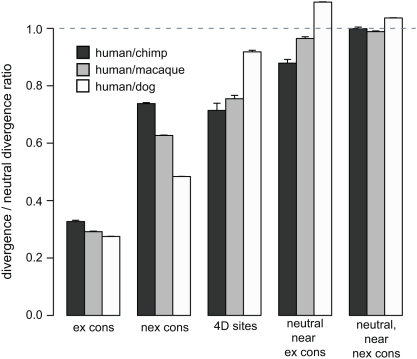
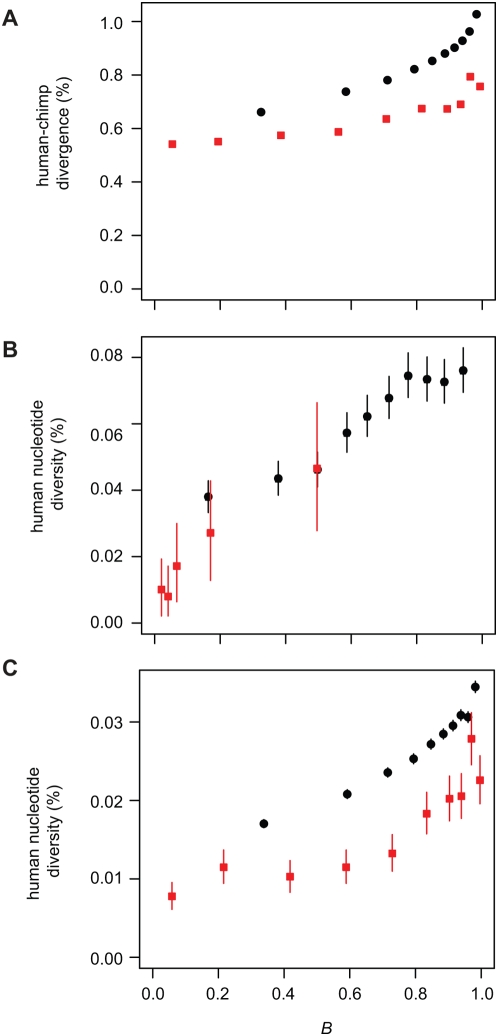
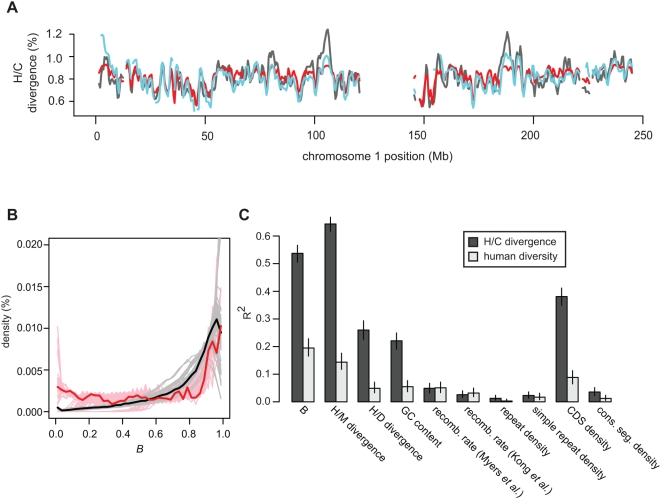
Selection acting on genomic functional elements can be detected by its indirect effects on population diversity at linked neutral sites. To illuminate the selective forces that shaped hominid evolution, we analyzed the genomic distributions of human polymorphisms and sequence differences among five primate species relative to the locations of conserved sequence features. Neutral sequence diversity in human and ancestral hominid populations is substantially reduced near such features, resulting in a surprisingly large genome average diversity reduction due to selection of 19-26% on the autosomes and 12-40% on the X chromosome. The overall trends are broadly consistent with "background selection" or hitchhiking in ancestral populations acting to remove deleterious variants. Average selection is much stronger on exonic (both protein-coding and untranslated) conserved features than non-exonic features. Long term selection, rather than complex speciation scenarios, explains the large intragenomic variation in human/chimpanzee divergence. Our analyses reveal a dominant role for selection in shaping genomic diversity and divergence patterns, clarify hominid evolution, and provide a baseline for investigating specific selective events.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
