

Experimentally based contact energies decode interactions responsible for protein-DNA affinity and the role of molecular waters at the binding interface
- PMID: 19429892
- PMCID: PMC2709573
- DOI: 10.1093/nar/gkp289
Experimentally based contact energies decode interactions responsible for protein-DNA affinity and the role of molecular waters at the binding interface
Abstract
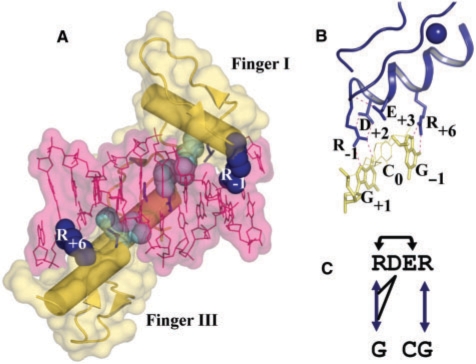
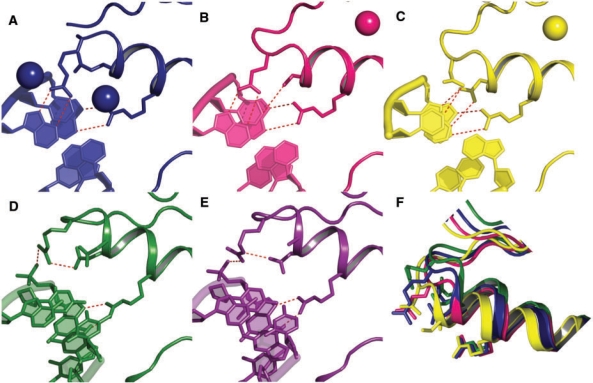
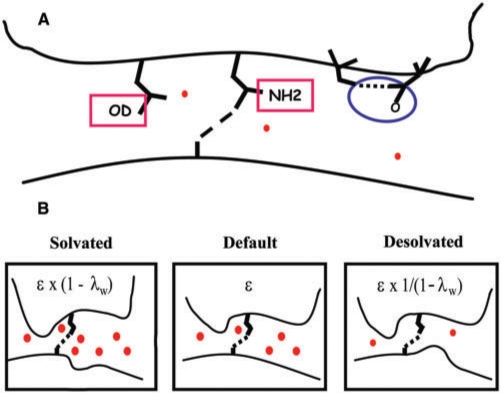
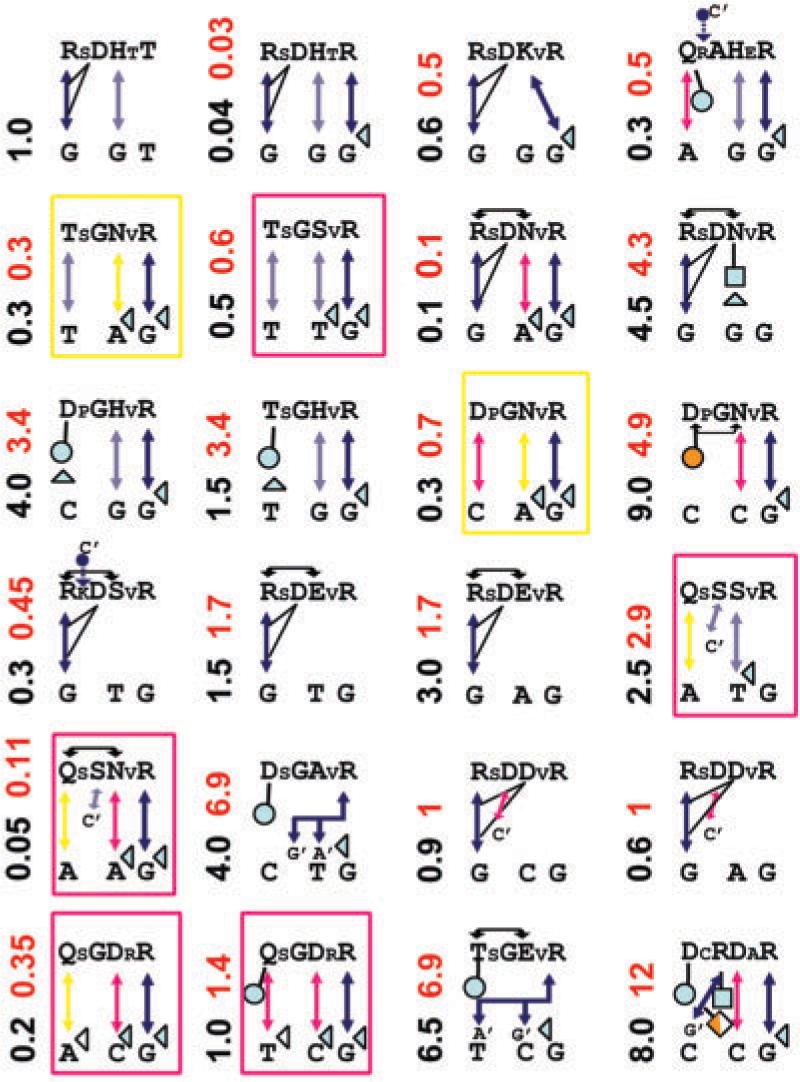
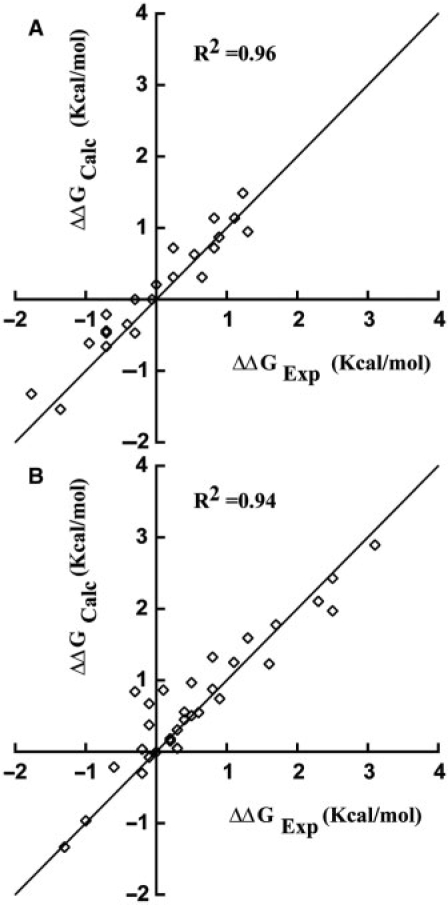
A major obstacle towards understanding the molecular basis of transcriptional regulation is the lack of a recognition code for protein-DNA interactions. Using high-quality crystal structures and binding data on the promiscuous family of C(2)H(2) zinc fingers (ZF), we decode 10 fundamental specific interactions responsible for protein-DNA recognition. The interactions include five hydrogen bond types, three atomic desolvation penalties, a favorable non-polar energy, and a novel water accessibility factor. We apply this code to three large datasets containing a total of 89 C(2)H(2) transcription factor (TF) mutants on the three ZFs of EGR. Guided by molecular dynamics simulations of individual ZFs, we map the interactions into homology models that embody all feasible intra- and intermolecular bonds, selecting for each sequence the structure with the lowest free energy. These interactions reproduce the change in affinity of 35 mutants of finger I (R(2) = 0.998), 23 mutants of finger II (R(2) = 0.96) and 31 finger III human domains (R(2) = 0.94). Our findings reveal recognition rules that depend on DNA sequence/structure, molecular water at the interface and induced fit of the C(2)H(2) TFs. Collectively, our method provides the first robust framework to decode the molecular basis of TFs binding to DNA.
Figures




References
-
- Siggia ED. Computational methods for transcriptional regulation. Curr. Opin. Genet. Dev. 2005;15:214–221. - PubMed
-
- Ladomery M, Dellaire G. Multifunctional zinc finger proteins in development and disease. Ann. Hum. Genet. 2002;66:331–342. - PubMed
-
- Benos PV, Lapedes AS, Stormo GD. Probabilistic code for DNA recognition by proteins of the EGR family. J. Mol. Biol. 2002;323:701–727. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
