

Text-based over-representation analysis of microarray gene lists with annotation bias
- PMID: 19429895
- PMCID: PMC2699530
- DOI: 10.1093/nar/gkp310
Text-based over-representation analysis of microarray gene lists with annotation bias
Abstract
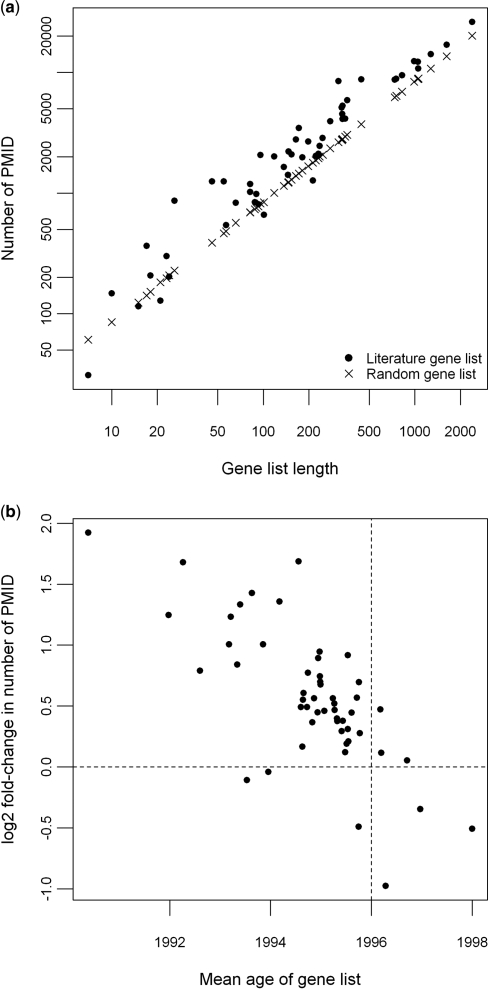
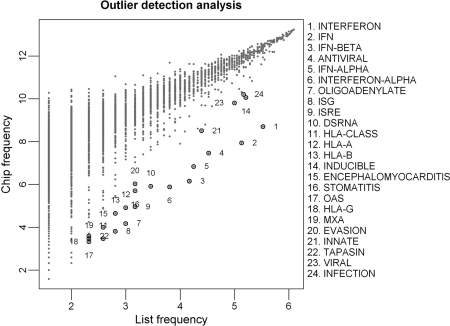
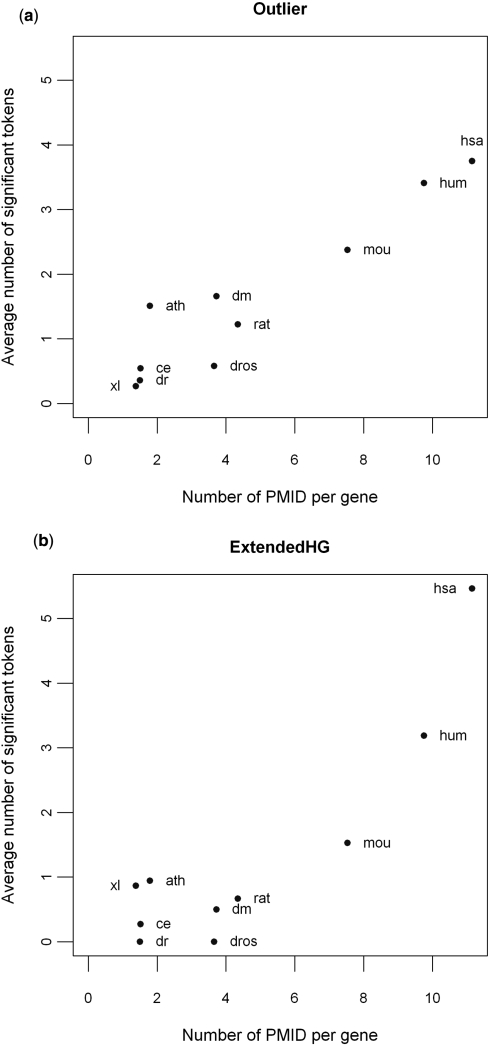
A major challenge in microarray data analysis is the functional interpretation of gene lists. A common approach to address this is over-representation analysis (ORA), which uses the hypergeometric test (or its variants) to evaluate whether a particular functionally defined group of genes is represented more than expected by chance within a gene list. Existing applications of ORA have been largely limited to pre-defined terminologies such as GO and KEGG. We report our explorations of whether ORA can be applied to a wider mining of free-text. We found that a hitherto underappreciated feature of experimentally derived gene lists is that the constituents have substantially more annotation associated with them, as they have been researched upon for a longer period of time. This bias, a result of patterns of research activity within the biomedical community, is a major problem for classical hypergeometric test-based ORA approaches, which cannot account for such bias. We have therefore developed three approaches to overcome this bias, and demonstrate their usability in a wide range of published datasets covering different species. A comparison with existing tools that use GO terms suggests that mining PubMed abstracts can reveal additional biological insight that may not be possible by mining pre-defined ontologies alone.
Figures
References
-
- Al Shahrour F, Diaz-Uriarte R, Dopazo J. FatiGO: a web tool for finding significant associations of Gene Ontology terms with groups of genes. Bioinformatics. 2004;20:578–580. - PubMed
-
- Dahlquist KD, Salomonis N, Vranizan K, Lawlor SC, Conklin BR. GenMAPP, a new tool for viewing and analyzing microarray data on biological pathways. Nat Genet. 2002;31:19–20. - PubMed
