

doi: 10.1038/nbt0509-455.
How to map billions of short reads onto genomes
Affiliations
- PMID: 19430453
- PMCID: PMC2836519
- DOI: 10.1038/nbt0509-455
Item in Clipboard
How to map billions of short reads onto genomes
Nat Biotechnol.
2009 May.
Abstract
Mapping the vast quantities of short sequence fragments produced by next-generation sequencing platforms is a challenge. What programs are available and how do they work?
Figures
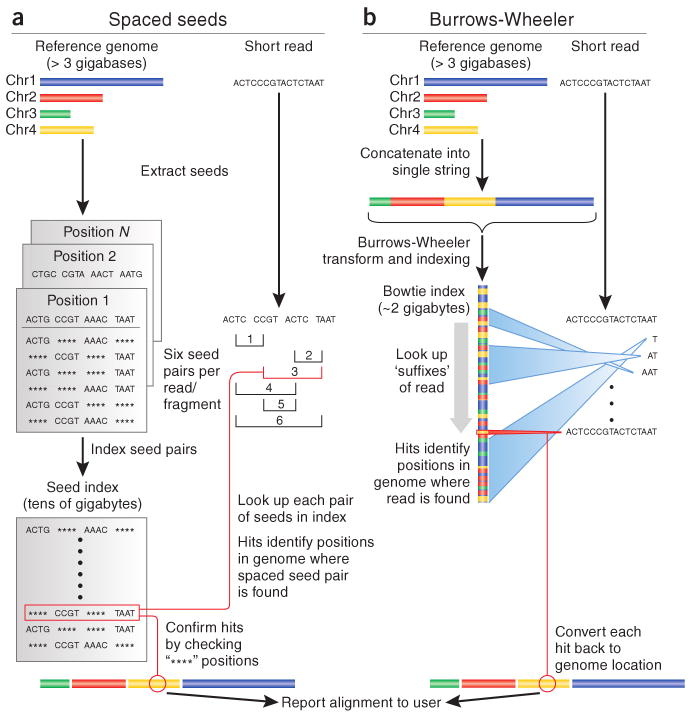
Two recent algorithmic approaches for aligning short (20–200-bp) sequencing reads. (a) Algorithms based on spaced-seed indexing, such as Maq, index the reads as follows: each position in the reference is cut into equal-sized pieces, called ‘seeds’ and these seeds are paired and stored in a lookup table. Each read is also cut up according to this scheme, and pairs of seeds are used as keys to look up matching positions in the reference. Because seed indices can be very large, some algorithms (including Maq) index the reads in batches and treat substrings of the reference as queries. (b) Algorithms based on the Burrows-Wheeler transform, such as Bowtie, store a memory-efficient representation of the reference genome. Reads are aligned character by character from right to left against the transformed string. With each new character, the algorithm updates an interval (indicated by blue ‘beams’) in the transformed string. When all characters in the read have been processed, alignments are represented by any positions within the interval. Burrows-Wheeler–based algorithms can run substantially faster than spaced seed approaches, primarily owing to the memory efficiency of the Burrows-Wheeler search. Chr., chromosome.

RNA-Seq assays produce short reads sequenced from processed mRNAs. Aligning these reads to the genome with Bowtie or Maq will produce the alignments shown in black but will fail to align the blue reads. A spliced-read mapper such as TopHat or ERANGE will also report the (blue) alignments spanning intron boundaries.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
