

Genome size in Hieracium subgenus Hieracium (Asteraceae) is strongly correlated with major phylogenetic groups
- PMID: 19433417
- PMCID: PMC2706716
- DOI: 10.1093/aob/mcp107
Genome size in Hieracium subgenus Hieracium (Asteraceae) is strongly correlated with major phylogenetic groups
Abstract
Background and aims: Hieracium subgenus Hieracium is one of the taxonomically most intricate groups of vascular plants, due to polyploidy and a diversity of breeeding systems (sexuality vs. apomixis). The aim of the present study was to analyse nuclear genome size in a phylogenetic framework and to assess relationships between genome size and ploidy, breeding system and selected ecogeographic features.
Methods: Holoploid and monoploid genome sizes (C- and Cx-values) of 215 cultivated plants from 89 field populations of 42 so-called 'basic' Hieracium species were determined using propidium iodide flow cytometry. Chromosome counts were available for all analysed plants, and all plants were tested experimentally for their mode of reproduction (sexuality vs. apomixis). For constructing molecular phylogenetic trees, the external transcribed spacer region of nuclear ribosomal DNA was used.
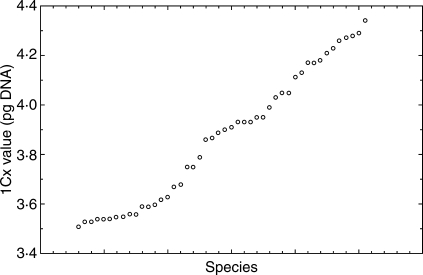
Key results: The mean 2C values differed up to 2.37-fold among different species (from 7.03 pg in diploid to 16.67 in tetraploid accessions). The 1Cx values varied 1.22-fold (between 3.51 and 4.34 pg). Variation in 1Cx values between conspecific (species in a broad sense) accessions ranged from 0.24% to 7.2%. Little variation (not exceeding the approximate measurement inaccurracy threshold of 3.5%) was found in 33 species, whereas variation higher than 3.5% was detected in seven species. Most of the latter may have a polytopic origin. Mean 1Cx values of the three cytotypes (2n, 3n and 4n) differed significantly (average of 3.93 pg in diploids, 3.82 pg in triploids and 3.78 pg in tetraploids) indicating downsizing of genomes in polyploids. The pattern of genome size variation correlated well with two major phylogenetic clades which were composed of species with western or eastern European origin. The monoploid genome size in the 'western' species was significantly lower than in the 'eastern' ones. Correlation of genome size with latitude, altitude and selected ecological characters (light and temperature) was not significant. A longitudinal component was only apparent for the whole data set, but absent within the major lineages.
Conclusions: Phylogeny was the most important factor explaining the pattern of genome size variation in Hieracium sensu stricto, species of western European origin having significantly lower genome size in comparison with those of eastern European origin. Any correlation with ecogeographic variables, including longitude, was outweighed by the divergence of the genus into two major phylogenetic lineages.
Figures






References
-
- Arnheim N. Concerted evolution of multigene families. In: Nei M, Koehn R, editors. Evolution of genes and proteins. Sunderland, MA: Sinauer; 1983. pp. 38–61.
-
- Arumuganathan K, Earle ED. Nuclear DNA content of some important plant species. Plant Molecular Biology Reports. 1991;9:208–218.
-
- Bancheva S, Greilhuber J. Genome size in Bulgarian Centaurea s.l. (Asteraceae) Plant Systematics and Evolution. 2006;257:95–117.
