

Detecting gene-gene interactions that underlie human diseases
- PMID: 19434077
- PMCID: PMC2872761
- DOI: 10.1038/nrg2579
Detecting gene-gene interactions that underlie human diseases
Abstract
Following the identification of several disease-associated polymorphisms by genome-wide association (GWA) analysis, interest is now focusing on the detection of effects that, owing to their interaction with other genetic or environmental factors, might not be identified by using standard single-locus tests. In addition to increasing the power to detect associations, it is hoped that detecting interactions between loci will allow us to elucidate the biological and biochemical pathways that underpin disease. Here I provide a critical survey of the methods and related software packages currently used to detect the interactions between genetic loci that contribute to human genetic disease. I also discuss the difficulties in determining the biological relevance of statistical interactions.
Figures
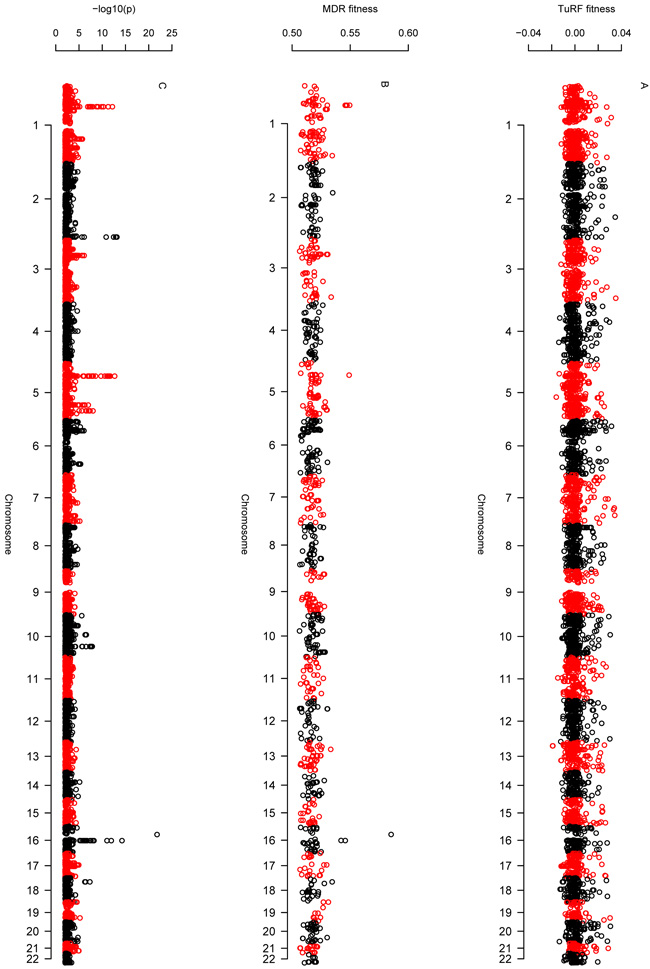
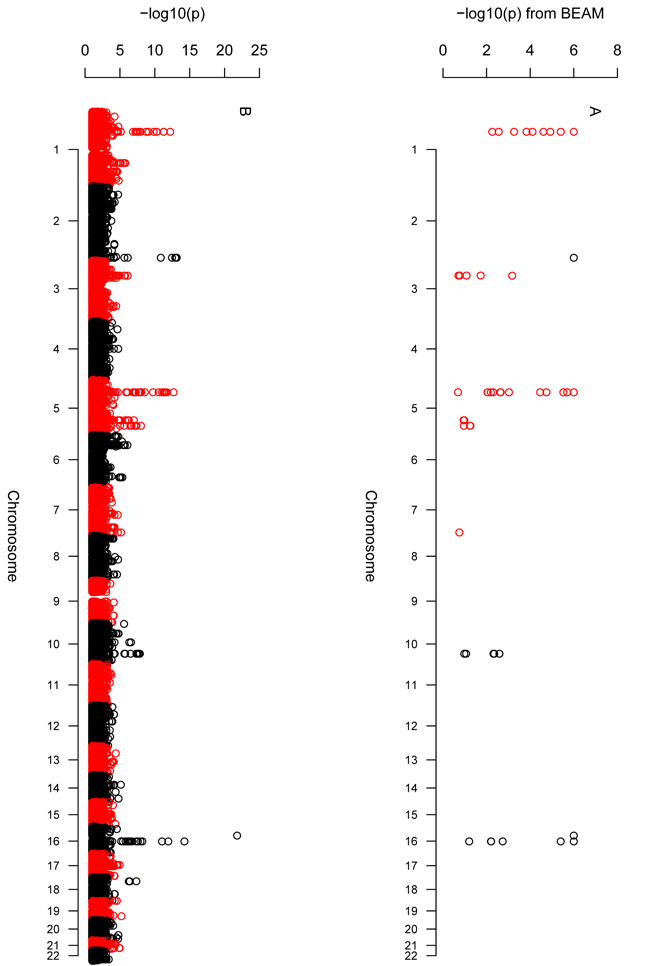
Comment in
-
Empirical tests for compositional epistasis.Nat Rev Genet. 2010 Feb;11(2):166. doi: 10.1038/nrg2579-c1. Nat Rev Genet. 2010. PMID: 20084088 Free PMC article. No abstract available.
-
Statistical interaction in human genetics: how should we model it if we are looking for biological interaction?Nat Rev Genet. 2011 Jan;12(1):74. doi: 10.1038/nrg2579-c2. Epub 2010 Nov 23. Nat Rev Genet. 2011. PMID: 21102529 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
