Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays
- PMID: 19435513
- PMCID: PMC2686739
- DOI: 10.1186/1471-2164-10-221
Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays
Abstract
Background: High-throughput cDNA synthesis and sequencing of poly(A)-enriched RNA is rapidly emerging as a technology competing to replace microarrays as a quantitative platform for measuring gene expression.
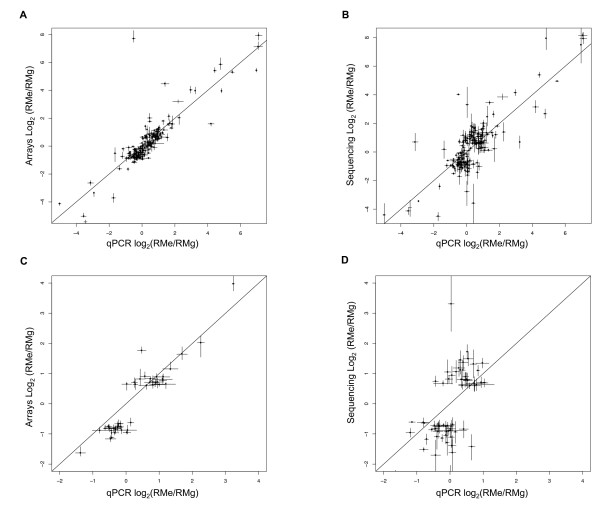
Results: Consequently, we compared full length cDNA sequencing to 2-channel gene expression microarrays in the context of measuring differential gene expression. Because of its comparable cost to a gene expression microarray, our study focused on the data obtainable from a single lane of an Illumina 1 G sequencer. We compared sequencing data to a highly replicated microarray experiment profiling two divergent strains of S. cerevisiae.
Conclusion: Using a large number of quantitative PCR (qPCR) assays, more than previous studies, we found that neither technology is decisively better at measuring differential gene expression. Further, we report sequencing results from a diploid hybrid of two strains of S. cerevisiae that indicate full length cDNA sequencing can discover heterozygosity and measure quantitative allele-specific expression simultaneously.
Figures