

Berkeley PHOG: PhyloFacts orthology group prediction web server
- PMID: 19435885
- PMCID: PMC2703887
- DOI: 10.1093/nar/gkp373
Berkeley PHOG: PhyloFacts orthology group prediction web server
Abstract
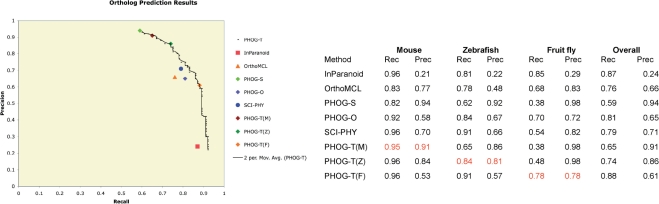
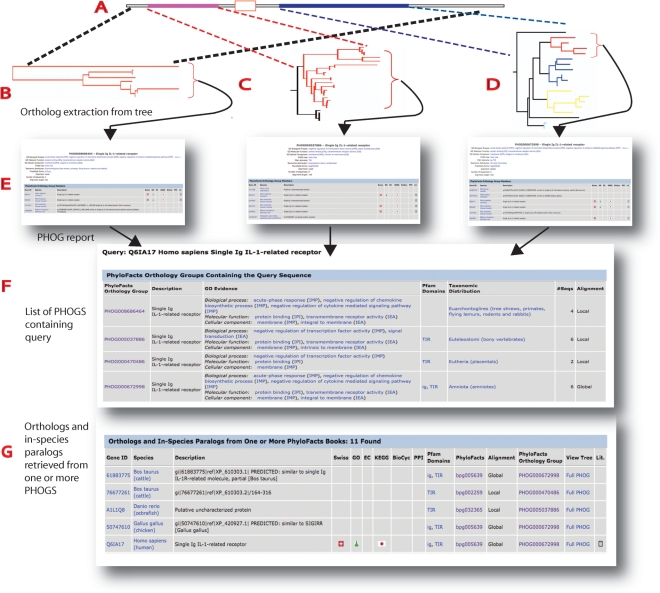
Ortholog detection is essential in functional annotation of genomes, with applications to phylogenetic tree construction, prediction of protein-protein interaction and other bioinformatics tasks. We present here the PHOG web server employing a novel algorithm to identify orthologs based on phylogenetic analysis. Results on a benchmark dataset from the TreeFam-A manually curated orthology database show that PHOG provides a combination of high recall and precision competitive with both InParanoid and OrthoMCL, and allows users to target different taxonomic distances and precision levels through the use of tree-distance thresholds. For instance, OrthoMCL-DB achieved 76% recall and 66% precision on this dataset; at a slightly higher precision (68%) PHOG achieves 10% higher recall (86%). InParanoid achieved 87% recall at 24% precision on this dataset, while a PHOG variant designed for high recall achieves 88% recall at 61% precision, increasing precision by 37% over InParanoid. PHOG is based on pre-computed trees in the PhyloFacts resource, and contains over 366 K orthology groups with a minimum of three species. Predicted orthologs are linked to GO annotations, pathway information and biological literature. The PHOG web server is available at http://phylofacts.berkeley.edu/orthologs/.
Figures
References
-
- Sjölander K. Phylogenomic inference of protein molecular function: advances and challenges. Bioinformatics. 2004;20:170–179. - PubMed
-
- Fitch WM. Distinguishing homologous from analogous proteins. Syst. Zool. 1970;19:99–113. - PubMed
-
- Eisen JA. Phylogenomics: improving functional predictions for uncharacterized genes by evolutionary analysis. Genome Res. 1998;8:163–167. - PubMed
-
- Friedberg I. Automated protein function prediction—the genomic challenge. Brief Bioinform. 2006;7:225–242. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
